ОСНОВИ МЕДИЧНОЇ БІОЛОГІЇ - 2012
Хромосомні хвороби. Цитогенетичний метод їх дослідження. Спадкові хвороби, їх класифікація
Залежно від ролі спадковості і середовища всі хвороби можна розділити на три групи: 1) спадкові хвороби; 2) хвороби зі спадковою схильністю (мультифакторіальні) і 3) неспадкові хвороби. Спадкові хвороби — хвороби, причиною яких є генна, хромосомна або геномна мутація. Прояв патологічної дії мутації практично не залежить від середовища. Останнє може лише міняти вираженість симптомів і тяжкість їх перебігу. Хвороби зі спадковою схильністю розвиваються в людей з певним генотипом під дією факторів середовища. Неспадкові хвороби - хвороби, причиною яких є фактори зовнішнього середовища (травми, опіки, інфекційні хвороби). Але і при цих захворюваннях спадковість впливає на перебіг патологічного процесу.
Термін "спадкові хвороби" слід відрізняти від термінів "уроджені хвороби" і "сімейні хвороби". Вони не є синонімами. Уроджені - всі хвороби, які наявні вже при народженні дитини, а зумовлені вони можуть бути як спадковими, так і неспадковими факторами. Спадкові хвороби - гемофілія, ахондроплазія (вкорочення довгих трубчастих кісток), які проявляються вже при народженні, - уроджені хвороби. Спадкові хвороби - міопатії, прогерія (передчасне старіння), які проявляються в юнацькому віці, хорея Гентингтона (середній вік початку якої становить 38-40 років), не є уродженими в повному розуміння цього поняття. Сімейні - всі хвороби (спадкові і неспадкові), які зустрічаються в членів однієї сім'ї. Неспадкові сімейні хвороби виникають під впливом однакового шкідливого фактора, який діє в цій сім'ї (наприклад, професійна шкідливість).
Розрізняють генетичну і клінічну класифікацію спадкових хвороб. В основу генетичної класифікації покладений етіологічний принцип, а саме: тип мутації і характер взаємодії із середовищем. Всю спадкову патологію можна розділити на 5 груп: 1) генні хвороби; 2) хромосомні хвороби; 3) хвороби зі спадковою схильністю (мультифакторіальні); 4) генетичні хвороби соматичних клітин; 5) хвороби генетичної несумісності матері і плода. Власне спадкові хвороби поділяються на дві великі групи: генні і хромосомні.
Генні хвороби - хвороби, зумовлені генними мутаціями. Вони передаються в ряді поколінь за законами Г. Менделя. Хромосомні хвороби зумовлені хромосомними і геномними мутаціями. Більшість хромосомних хвороб, зумовлених анеуплодією, взагалі не успадковується (летальний ефект), а структурні перебудови хромосом передаються з додатковими перекомбінаціями, що виникають у мейозі носія аберації. Хвороби із спадковою схильністю можуть бути моногенними і полігенними. Для їх реалізації необхідна не лише відповідна генетична конституція індивідуума, але і фактор або комплекс факторів середовища, які відіграють роль пускових моментів у формування патології. Генетичні хвороби соматичних клітин пов'язані з виникненням при онкологічних новоутвореннях у соматичних клітинах специфічних хромосомних аберацій, які викликають активацію онкогенів. До цих хвороб відносять ретинобластому, пухлину Вільмса (рак нирок). Хвороби генетичної несумісності матері і плода розвиваються в результаті імунної реакції матері на антигени плода (гемолітична хвороба новонароджених).
Спадкова патологія настільки різноманітна, що зустрічається в практиці медика будь-якої спеціальності. Відповідно до медичних спеціальностей існує клінічна класифікація спадкових хвороб. В її основі лежить системний і органний принцип. Розрізняють: спадкові хвороби нервової системи; спадкові хвороби внутрішніх органів; спадкові хвороби шкіри; спадкові хвороби очей, інших органів. Дуже мало спадкових хвороб, при яких вибірково уражається лише одна система. Більшість спадкових хвороб проявляється у вигляді комплексу патологічних ознак - синдромів(синдром Дауна, синдром Клайнфельтера). Принципи діагностики спадкових хвороб ґрунтуються на даних клінічного діагнозу, який уточнюється за допомогою генетичних методів при проведенні медико-генетичного консультування.
Необхідно підкреслити суть визначення поняття спадкові хвороби: основним тут є не факт спадкування хвороби (хоча це має місце для багатьох з них), а те, що причина хвороби - порушення у спадковому (генетичному) апараті клітин обох або одного із батьків.
Хромосомні хвороби
До хромосомних хвороб відносять різні форми патологій, які клінічно проявляються багатьма вадами розвитку, генетичною основою яких є хромосомні (зміна структури хромосом) або геномні (зміна числа хромосом) мутації.
Більшість хромосомних хвороб виникають зазвичай внаслідок нових мутацій і не успадковуються в поколіннях. Фенотипову основу хромосомних хвороб визначають порушення раннього ембріонального розвитку, тому патологічні зміни, які утворюються вже в пренатальному періоді розвитку зумовлюють елімінацію ембріону, плоду або визначають клінічну картину захворювання вже у новонародженого (за винятком деяких порушень статевого розвитку, які з’являються під час статевого дозрівання).
Деякі хромосомні хвороби, які найчастіше зустрічаються були описані давно, як клінічні синдроми порушень розвитку, раніше ніж був встановлений їхній зв’язок зі змінами в хромосомах. Це хвороба Дауна (1866р.), синдром Клайнфельтера (1942р.), синдром Шерешевського - Тернера (1925, 1938р.р.). Встановлення зв’язку міх хворобами і змінами кількості хромосом було доведено тільки у 1959р. На даний час встановлено вже більше 500 хромосомних хвороб - порушень кількості та структури хромосом.
Роль хромосомної патології значна в пренатальній смерті ембріонів та плодів (40%), близько 6% мертвонароджених мають хромосомні порушення. На 1000 новонароджених 3-4 мають хромосомну патологію, серед дітей з природженими вадами розвитку близько 40% мають хромосомні порушення.
Виникнення хромосомних хвороб пов'язане з порушеннями розходження хромосом під час першого та другого поділів анафази мейозу, а також з виникненням хромосомних аберацій (делеціями, дуплікаціями, транслокаціями та іншими порушеннями структури). Хромосомні хвороби виникають заново внаслідок мутацій в гаметах одного із здорових батьків або в зиготі на перших стадіях дроблення. Якщо мутація виникла в гаметах, це повна форма хвороби, якщо на стадії дроблення зиготи - мозаїчна форма хвороби, при якій одні клітини матимуть нормальний каріотип, а інші - мутаційний. Організми - мозаїки можуть мати 2-3 або більшу кількість клітинних клонів. Патологія визначається кількістю змінених клітин та характеру мутації (за аутосомами, гоносомами, або часткові моносомії чи трисомії). При повній формі зміни хромосом наявні в усіх клітинах нащадка. На відміну від генних, хромосомні мутації охоплюють значно більший об'єм генетичного матеріалу і характеризуються множинними ураженнями, які проявляються летальністю і природженими вадами розвитку. Хворі на хромосомні хвороби займають майже 25% ліжкового фонду всього світу. Для діагностики хромосомних хвороб застосовують цитогенетичний метод. Кількісні і структурні порушення хромосом видно під мікроскопом.
Геномні мутації, які пов'язані зі збільшенням або зменшенням гаплоїдних наборів хромосом, несумісні з життям людини. У клініці зустрічаються лише гетероплоїдії - трисоміії, рідше тетра- і пентасомії, один варіант моносомії, нулісомія несумісна з життям. Розрізняють хромосомні хвороби, зумовлені зміною числа автосом, і хромосомні хвороби, пов'язані з порушенням числа статевих хромосом. Хромосомні хвороби, зумовлені зміною числа аутосом: синдром Дауна, синдром Едвардса, синдром Патау.
Синдром Дауна (трисомія - 21). Клінічну картину синдрому вперше в 1866 р. описав англійський лікар Л. Даун, назвавши захворювання "монголоїдною ідіотією". У 1959 р. французький учений І. Лежен виявив у каріотипі хворих зайву хромосому 21. Каріотипи хворих - 47, XX, +21 або 47, XY, +21. Частота 1:1100, а в деяких регіонах -1:700-1:800 новонароджених. Ризик народження дітей з синдромом Дауна зростає з віком матері. На частоту їх народження не впливають статеві, расові, географічні і популяційні відмінності. Комплекс природжених вад розвитку, характерних для синдрому Дауна, зумовлює клінічну картину "всі діти з однієї сім'ї"".
Клінічні діагностичні ознаки: низький зріст, різні ступені розумової відсталості, черепно-лицеві аномалії: косий розріз очей, коротка шия, епікант (нависаюча складка шкіри біля внутрішнього кута ока), плоске обличчя, маленький короткий ніс, великий язик, маленькі деформовані вуха (рис.). Характерні також м'язова гіпотонія, розхитаність суглобів, поперечна складка на долонях, клинодактилія (викривлення) мізинця. Вроджені вади внутрішніх органів (серця), знижений імунітет часто є причиною смерті цих дітей.
Цитогенетичні варіанти синдрому різноманітні. Основну частку (94%) складають випадки повної трисомії 21 як результат нерозходження хромосом у мейозі. При цьому внесок материнського нерозходження складає 80% батьківського - 20%. Приблизно 4% хворих мають транслокаційну форму (транслокація хромосоми 21 найчастіше на хромосоми 13 або 22) і 2% - мозаїцизм внаслідок мітотичного нерозходження, коли одна частина клітин має нормальну кількість хромосом (46), а інша - анеуплоїдну (47). Транслокаційна форма не залежить від віку матері, тому є високий ризик повторного народження хворої дитини в сім'ї.
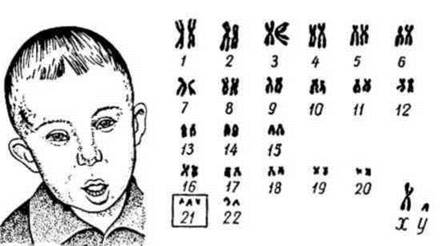
Рис. 33. Синдром Дауна (трисомія 21) і каріограма хворого.
Синдром Патау (трисомія-13). Каріотип 47, XX, +13 або 47, XY, +13. Частота 1:5000 - 1:7000 новонароджених. Клінічні діагностичні ознаки: щілини верхньої губи і піднебіння, зменшений об'єм черепа, перекошений, низький лоб, мікрофтальмія (малий розмір очного яблука), анофтальмія (відсутність одного або обох очних яблук), перенісся запале, деформовані вушні раковини, полідактилія, вроджені вади серця, інших внутрішніх органів. Вирішальним у діагностиці є цитогенетичне дослідження. Прогноз для життя при синдромі Патау несприятливий: більшість дітей вмирає в перші тижні або місяці. Середня тривалість життя 130 днів: 60% хворих помирають впродовж перших 3 місяців після народження, тільки близько 10% дітей живуть більше року.
Синдром Едвардса (трисомія-18). Каріотип 47, XX, +18 або 47, XY, +18. Частота 1:5000-1:7000. Співвідношення хлопчиків і дівчаток дорівнює 1:3. Причини переважання хворих дівчаток поки що невідомі. Клінічні діагностичні ознаки: доліхоцефалічний череп (переважання поздовжнього діаметра голови над поперечним), малі рот і нижня щелепа, очні щілини вузькі, вушні раковини деформовані, флексорне положення кистей, аномальна стопа ("стопа-качалка"). Для синдрому характерні вроджені вади серця, скелетної системи, нирок, статевих органів. Діти переважно вмирають до 2 місяців. Діагностика - цитогенетичне дослідження.
Хромосомні хвороби, зумовлені зміною структури аутосом - синдром «котячого крику».
Синдром «котячого крику» (синдром 5р- - делеція короткого плеча п ’ятої хромосоми). Частота даної патології серед новонароджених становить 1:50000. Співвідношення статей - хлопчики : дівчатка - 1:1,6. Основними фенотиповими ознаками синдрому є низька маса тіла при народженні (близько 2600 г), мікроцефалія, кругле, "місяцеподібне" обличчя на перших роках життя і вузьке обличчя в більш старшому віці, антимонголоїдний розтин очей, епікант, гіпертелоризм, косоокість, катаракта, осередки депігментації сітківки, атрофія зорових нервів, сплюснута спинка носа, високе піднебіння, у деяких хворих із щілиною; мікроретрогнатія. Вушні раковини деформовані і розташовані нижче звичайного, іноді з преартикулярною заглибиною. Часто відмічаються дефекти кістково-м'язової системи: клинодактилія мізинців рук, синдактилія пальців ніг, клишоногість, м'язова гіпотонія, розходження м'язів живота, пупкові та пахвинні грижі. Патогномонічним симптомом є своєрідний крик під час народження, що нагадує лемент кішки. Він присутній у дітей першого року життя і пов'язаний як із порушенням ЦНС, так і зі змінами гортані (зменшення надгортанника, звуження щілини гортані, набряк слизової оболонки). При синдромі 5р- зазвичай присутня глибока розумова відсталість (імбецильність та ідіотія), недорозвинення мови, виражена затримка фізичного і моторного розвитку, парези кінцівок. При патолого-анатомічних дослідженнях виявляють дифузну атрофію мозку, мозочка, гідроцефалію, рідше вади серця, нирок, легень, дисплазію тимуса.
Прогноз відносно життя при часткових трисоміях та моносоміях хромосоми 5 залежить від вираженості симптомів, більшість хворих доживають до підліткового віку.
Хромосомні хвороби, зумовлені зміною числа статевих хромосом - синдром Шерешевського-Тернера, синдром Клайнфельтера, синдром трипло-Х, синдром дисомії за Y-хромосомою.
Синдром Шерешевського-Тернера (моносомія -X). Каріотип 45, ХО. У клітинах відсутні тільця статевого хроматину. Частота 1:2000-1:5000. Синдром описали російський клініцист М.А. Шерешевський (1925) і Г. Тернер (1938). Клінічні діагностичні ознаки: синдром виявляється в жінок; низький зріст, коротка шия з надлишком шкіри і крилоподібними складками (шия сфінкса), низька межа росту волосся на потилиці, грудна клітка щитоподібної форми з широко розставленими сосками, недорозвинення яєчників. Вади серця діагностують майже у чверті хворих, найчастіше це коарктація аорти, стеноз легеневої артерії, незарощення міжшлуночкової перегородки, артеріальної протоки. Рідше можна знайти патологію нирок. У підлітковому віці дуже часто трапляється низький зріст (150-153 см), чоловічий тип статури. Прогноз для життя сприятливий. Нащадків хворі не мають, хоча опубліковані поодинокі спостереження за жінками з каріологічно підтвердженим синдромом Шерешевського-Тернера, які народили здорових дітей (Wray H.Z. et al., 1981).
Синдром Клайнфельтера. Каріотип 47, XXY. Частота 1:400. Синдром виявляється лише в осіб чоловічої статі переважно при статевому дозріванні. Клінічні діагностичні ознаки: високий зріст, довгі кінцівки, євнухоїдизм, гінекомастія (збільшення молочних залоз), відсутність сперматогенезу, недорозвинення статевих залоз. Тільця статевого хроматину виявляються в 80% випадків. Інколи хворі з синдромом Клайнфельтера мають 48 і 49 хромосом (48, XXXY; 49, XXXXY). Чим більше Х-хромосом у каріотипі, тим вища ймовірність розвитку розумової відсталості.
Синдром трипло-Х (трисомія-X). Каріотип 47, XXX. Переважна більшість таких жінок мають нормальний фізичний та розумовий розвиток і виявляються випадково при обстеженні. Лише в деяких з них порушена репродуктивна функція. Більшість жінок мають нормальну плодючість, хоча зростає ризик мимовільних викиднів і хромосомних аберацій у нащадків. У клітинах - по два тільця статевого хроматину. При збільшенні числа Х-хромосом наростає ступінь відхилення від норми. У жінок з тетра- і пентасомією описані розумова відсталість, черепно-лицеві аномалії, аномалії зубів, скелета і статевих органів. Однак жінки навіть з тетрасомією по X- хромосомі мають нащадків.
Синдром дисомії за Y-хромосомою. Каріотип 47, XYY Частота 1:1000. Синдром виявляється в осіб чоловічої статі. За своїм розумовим і фізичним розвитком такі чоловіки не відрізняються від нормальних осіб. Помітних відхилень у статевому і гормональному статусі не виявлено. Проте деякі клініцисти вказували на підвищений ступінь агресивності в окремих з них.
Цитогенетичний метод
Основний метод визначення хромосомних хвороб — цитогенетичний, який включає: а) каріотипування (встановлення та аналіз каріотипу); б) диференційне забарвлення хромосом; в) визначення статевого хроматину (Y-хроматину, Х-хроматину).
Це метод генетики людини, який ґрунтується на вивченні хромосом людини за допомогою мікроскопа. Метод застосовується для: 1) вивчення каріотипів; 2) діагностики хромосомних хвороб людини; 3) складання карт хромосом; 4) вивчення мутаційного процесу в людських популяціях; 5) розв'язання деяких еволюційних питань.
Вивчення каріотипу людини (каріотипування). З метою вивчення каріотипу людини досліджують мітотичні (метафазні), рідше - мейотичні (профазні і метафазні) хромосоми. Існують прямий і непрямий методи їх вивчення. При прямому методі свіжий біопсійний матеріал досліджують відразу після одержання (кістковий мозок, пухлини, ембріональні тканини, хоріон, клітини гонад), при непрямому - після попереднього культивування на поживних середовищах. У даний час каріотип людини найчастіше досліджують у культурі лейкоцитів периферичної крові. Одержують препарати метафазних пластинок.
Основним методом ідентифікації хромосом людини продовжує залишатися метод їх систематизації шляхом вирізання кожної хромосоми з мікрофотографії метафазної пластинки і наклеювання їх на папері згідно з Денверською класифікацією. Хромосоми розподіляють на 7 груп, у межах яких встановлюється пара гомологічних хромосом. Так одержують ідіограму (каріограму) хромосом. У основу Денверської системи покладені особливості розмірів хромосом і розташування первинної перетяжки.
Проте ідентифікація хромосом тільки за цими показниками зустрічає великі труднощі. Фактично вдається встановити, до якої групи відноситься хромосома, а у межах групи визначити її місце і номер досить складно. На Паризькій конференції по стандартизації хромосом людини (1971р.) шведський генетик Т. Касперссоном (1969р.) запропонував методику диференційного забарвлення хромосом, яка значно підвищила можливості, дозволила точно визначити гомологічні хромосоми та порушення структури хромосом - хромосомні аберації (методами флуоресцентного забарвлення хромосом акрихін-іпритом та його похідними).
Визначення статевого хроматину. Розрізняють X- та Y-статевий хроматин (с). Х-статевий хроматин (тільце Барра) - це одна з двох Х-хромосом жінки, яка гетерохроматизується на ранніх стадіях ембріогенезу і переходить у генетично неактивний стан, що знижує дозу генів. Найчастіше Х-хроматин досліджують в ядрах клітин епітелію слизової оболонки ротової порожнини, де він має вигляд напівсферичної грудочки гетерохроматину, яка прилягає до внутрішньої мембрани ядра. Статевий хроматин виявляють також у нейтрофільних лейкоцитах у вигляді барабанної палички, що виступає на поверхні одного з сегментів ядра. ( рис.) Кількість грудочок статевого хроматину (або барабанних паличок) на одиницю менша від кількості Х-хромосом у соматичних клітинах, що виражається формулою: а = n—1, де а - кількість грудочок Х-хроматину, n - кількість X-хромосом. У здорової жінки (XX) 60-70% соматичних клітин містять одне тільце Барра, при трисомії за X-хромосомою (XXX) - два тільця Барра, при моносомії за Х-хромосомою (Х0) тільця Барра відсутні. У більшості здорових чоловіків (XY) в ядрах соматичних клітин немає жодного тільця Барра, однак у сучасних популяціях можуть зустрічатися до 8 %.
Y-хроматин (F-тільце) - це Y-хромосома в ядрах соматичних клітин чоловіків. Для виявлення Y-хроматину соматичні клітини забарвлюють флуорохромами і досліджують в люмінесцентному мікроскопі. Y-хромосома відрізняється від усіх інших хромосом інтенсивним зеленим світінням свого довгого плеча. В ядрі соматичної клітини здорового чоловіка видно одну ділянку флуоресценції, при полісомії Y (XYY) - дві і т. д.
Метод визначення статевого хроматину застосовують у наступних ситуаціях для: 1) визначення статі у випадку гермафродитизму; 2) визначення статі до народження дитини (при амніоцентезі); 3) діагностики спадкових хвороб, зумовлених дисбалансом статевих хромосом; 4) встановлення спадкової патології дітей з порушенням інтелекту; 5) виявлення спадкової патології при первинній і вторинній аменореї у жінок; 6) при безплідності в чоловіків і жінок; 7) у судово-медичній практиці (за дослідженнями плям крові можна встановити стать людини, якій належала ця кров).
Значення: визначення статевого хроматину дозволяє проводити без повного каріотипування, що потребує багато часу, експрес-діагностику числа статевих хромосом.
Зв'язок між кількістю Х-хромосом, кількістю тілець Барра в соматичних клітинам слизової оболонки порожнини ро ї а (А), кількістю "барабанних паличок" в ядрах лейкоцитів (Б)

Рис. 34. Визначення статевого Х-хроматину.
Цитогенетичний метод дозволяє встановити: а) кількість хромосом у каріотипі; б) генетичну стать організму; в) наявність хромосомних аберацій та їх локалізацію.
Застосування цитогенетичного методу в МГК дає можливість своєчасно діагностувати хромосомну патологію та попереджувати народження дитини з хромосомними синдромами.
Крім того, для діагностики хромосомних хвороб, можуть використовуватися біохімічні методи, так як при цих хворобах є ефект «дози генів», внаслідок чого змінюється концентрація деяких ферментів. Наприклад, при трисомії 21 (синдром Дауна) збільшується в 1,5 рази активність ферментів супероксидисмутази. Ефект дози встановлений для 30 генів, локалізованих в різних хромосомах.
Значення для діагностики хромосомної патології має фенотиповий аналіз (наприклад, портретна діагностика) та дерматогліфіка.