Молекулярная биология: Структура и функции белков - Степанов В.М. 2005
Химическое ;модифицирование белков
Типовые реакции химической модификации функциональных групп белка
9.2.1. є-Аминогруппы
ε-Аминогруппы принадлежат боковым цепям остатков лизина. Они располагаются, как правило, на поверхности глобулы, достаточно доступны и обладают химическими свойствами, характерными для алифатических первичных аминов. рКа этих групп обычно близок 10, поэтому реакции с этими группами проводят в слабощелочных средах при pH 8 и выше, т.е в условиях, когда они хотя бы частично депротонированы и могут выступать в качестве нуклеофильных агентов. Заметим, что а-аминогруппы реагируют сходным образом, однако на их реакционной способности сказывается несколько меньшая основность (рКа около 8).
Ацилирование. Эти группы легко ацилируются, для чего могут быть использованы многие реагенты, например активированные эфиры кислот:

Однако близкие по нуклеофильности фенолят-ионы остатков тирозина, как правило, вступают параллельно в ту же реакцию. Для более избирательного блокирования аминогрупп можно воспользоваться гораздо меньшей стабильностью к гидролизу сложных эфиров, получающихся при ацилировании фенольного гидроксила тирозина. Эта неустойчивость особенно значительна в случае моноэфиров дикарбоновых кислот, которые образуются при ацилировании тирозина ангидридами, например, янтарной или цитраконовой кислот. В этих случаях продукты модификации тирозина настолько нестабильны, что практически не обнаруживаются, так что удается достигнуть специфического ацилирования ε-аминогрупп лизина:
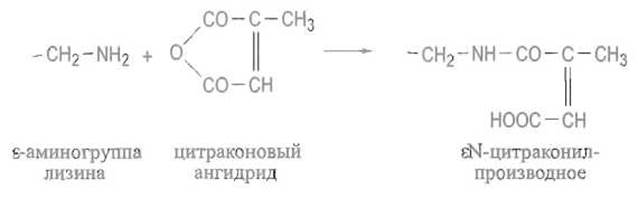
В полученном производном катионная в обычных условиях аминогруппа заменена анионным карбоксилат-ионом. Существенно, что продукты ацилирования аминогрупп лизина ангидридами дикарбоновых кислот вследствие сближенности высвободившейся при реакции карбоксильной группы с вновь образованной амидной связью лабильны и в кислой среде остатки дикарбоновых аминокислот отщепляются, высвобождая s-аминогруппы остатков лизина. Такую возможность обратимого блокирования ε-аминогрупп лизина используют для того, чтобы проводить гидролиз трипсином модифицированного, например цитраконилированного, белка сначала только по остаткам аргинина, а после разделения получающихся крупных фрагментов и деблокирования ε-аминогрупп — по остаткам лизина.
Арилирование. ε-Аминогруппы остатков лизина могут выступать в качестве нуклеофильных агентов в реакциях с 2,4-динитрофтор-бензолом и другими ароматическими активированными галоидными соединениями:
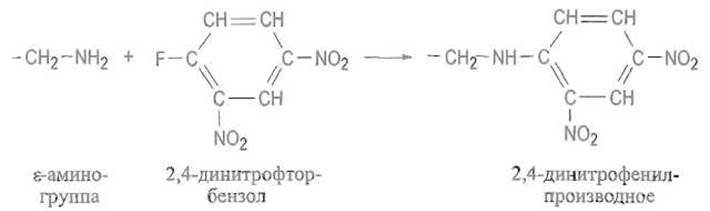
В образующемся динитрофенил-производном характерного желтого цвета (λmах ≈ 350+ 360 нм) азот аминогруппы полностью утрачивает основные свойства, а гидрофильная аминогруппа заменяется весьма гидрофобной структурой.
Образование оснований Шиффа. Аминогруппы реагируют с альдегидами, взаимодействуя с карбонильной группой в два этапа по следующей схеме:
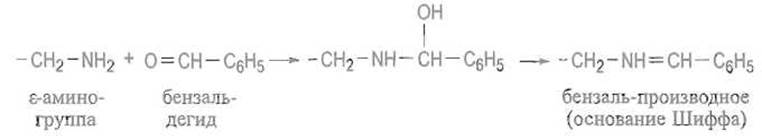
Эта реакция избирательна, однако основания Шиффа малоустойчивы и легко, особенно в кислой среде, расщепляются с регенерацией свободных аминогрупп и альдегида. Учитывая эго, прибегают к закреплению образовавшейся структуры путем восстановления двойной связи —CH=N— боргидридом натрия NaBН4:
![]()
Превращение в амидины. Модификация ε-аминогрупп лизина указанными реагентами приводит к изменению заряда в данной точке белковой молекулы, а иногда к введению гидрофобного заместителя. Реакция с имидоэфирами отличается тем, что в ее результате катионная аминогруппа замещается также катионной амидиновой — основность которой несколько выше, чем у аминов:
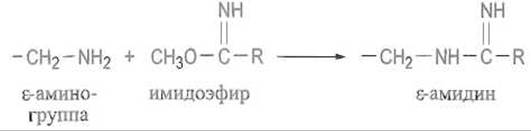
Если заместитель R невелик, то такая модификация приведет только к замене аминогруппы на амидиновую — ни заряд, ни гидрофидьность при этом заметно не изменятся, что позволит с большей уверенностью интерпретировать результаты модификации. По этим же соображениям бис-имидоэфиры охотно используют для соединения двух белковых молекул друг с другом. При обработке производного белка, модифицированного имидоэфиром по описанной схеме, большим избытком первичного амина, например метиламина, происходит перенос амидиновых групп на этот амин, остатки лизина высвобождаются и белок удается регенерировать.
9.2.2. Фенольные группы тирозина
Гидроксильные группы тирозина в белках располагаются частично внутри, частично на поверхности белковой глобулы. В последнем случае для них характерен рКа ≈ 10, соответствующий полупереходу:
![]()
Образующийся при этом фенолят-ион весьма активно вступает в реакции, свойственные также и ε-аминогруппам лизина, в частности в реакцию ацилирования.
Ацилирование. Эта реакция может проводиться различными реагентами, например ацетилимидазолом:
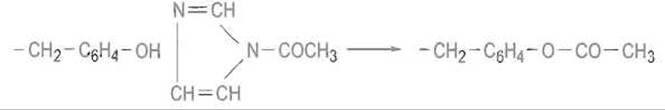
Гораздо характернее для тирозина, однако, реакции замещения в ароматическом ядре, рассматриваемые далее.
Иодирование. Эту реакцию особенно часто применяют как способ введения радиоактивных изотопов иода в белковую молекулу. Источником атомов иода может служить комплекс иода и йодистого калия; в некоторых случаях используют иод, образующийся при окислении иодид-иона в присутствии фермента пероксидазы. Последний способ делает реакцию более избирательной, так как образование свободного иода происходит там, куда может проникнуть крупная молекула пероксидазы. Иодирование приводит к образованию как моно-, так и дииодтирозина, причем замещение происходит в о-положении:

Результат введения иода в остаток тирозина неоднозначен: наряду с включением в структуру одного или двух объемистых атомов иода становится значительно более кислым гидроксил фенольной группы. Эту реакцию нередко используют для мечения белка радиоактивным иодом, причем модификации могут подвергаться различные остатки тирозина, локализованные на его поверхности.
Нитрование. Нитрование достигается действием тетранитрометана C(NO2)4, причем в роли активной частицы обычно выступает нитроний-катион +NO2:
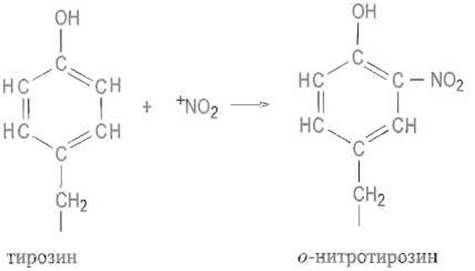
И в этом случае возможно, хотя и проходит со значительно большим трудом, введение второй нитрогруппы (также в о-положение).
Гидроксильная группа в о-нитротирозине, по существу, такая же, как в о-нитрофеноле, имеет выраженные кислотные свойства. Наряду с нитроний-катионами тетранитрометан образует и частицы со свойствами свободных радикалов +NC2. Они также реагируют с образованием о-нитротирозина, однако неспаренный электрон, делокализованный в ароматическом ядре, вызывает ряд побочных реакций, в частности димеризацию остатков о-нитротирозина.
Азосочетание. Реакция остатков тирозина с ароматическими солями диазония, также являющимися катионными реагентами, опять-таки приводит к образованию моно- и дио-замещенных продуктов с достаточно протяженной системой сопряженных двойных связей, что придаст им глубокое, обычно красно-оранжевое окрашивание (так называемая реакция Паули, иногда применяемая для обнаружения белков и пептидов, содержащих тирозин или гистидин):

Эта реакция также приводит к усилению кислотных свойств гидроксильной группы тирозина. Она достаточно специфична, затрагивая помимо тирозина лишь остатки гистидина. Однако иногда наблюдается реакция солей диазония с аминогруппами лизина, дающая чрезвычайно нестабильные триазены. Для аналитического определения о-нитро- и азотирозина иногда прибегают к восстановлению соответственно нитро- или азогрупп до аминогруппы. Получающийся при этом о-аминотирозин сохраняется при полном кислотном гидролизе белка и может быть определен с помощью аминокислотного анализатора.
9.2.3. Реакции карбоксильных групп
ω-Карбоксильные группы аспарагиновой и глутаминовой кислот, как правило, весьма многочисленны и в большинстве своем размещены в поверхностном слое белка. Как уже упоминалось, их реакционная способность существенным образом зависит от микроокружения. Наиболее часто применяют следующие методы модификации этих групп.
Реакция с аминами в присутствии карбодиимидов. Эту реакцию проводят в воде, используя водорастворимые карбодиимиды, например N-этил-N'-триметиламинопропил карбодиимид:
![]()
Реакция с этим или другими карбодиимидами протекает по следующей схеме (R1—N=C=N—R2 — карбодиимид, R3—NH2 — алифатический амин, часто метиловый эфир глицина):
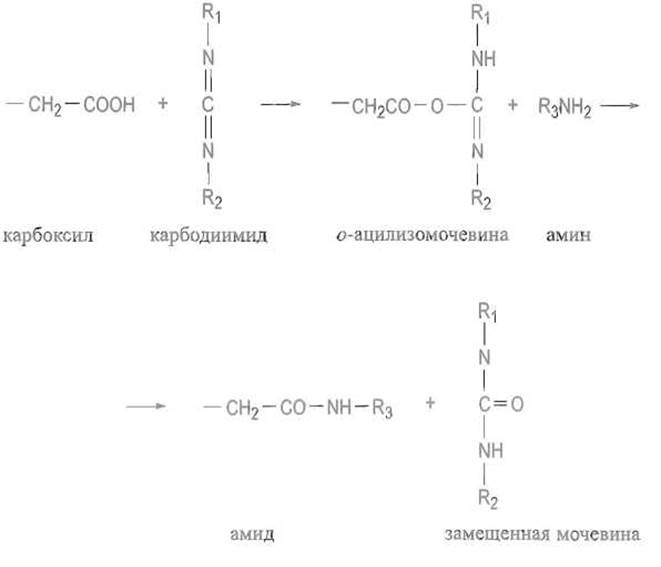
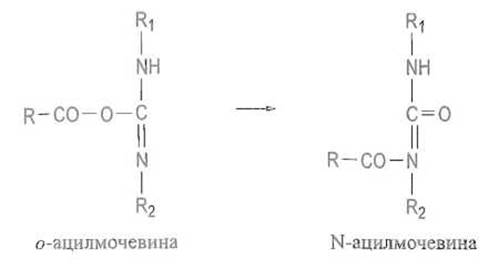
Если провести исчерпывающее замещение карбоксильных групп в белке, используя в качестве амина метиловый эфир глицина или, лучше, метиловый эфир небелковой аминокислоты, например норвалина, то по аминокислотному составу продукта реакции можно определить число карбоксильных групп, которые в нее вступили, и, следовательно, число доступных для реагента карбоксильных групп. Как правило, оно велико, и скрытыми оказываются лишь единичные группы. Следует иметь в виду, что продукт активации карбоксильной группы — о-ацилизомочевина — может не только вступать в реакцию с амином, но и стабилизироваться в результате внутримолекулярного переноса ацильного остатка с атомом кислорода на азот. Это приводит к образованию весьма стабильной N-ацилмочевины:
Реакции с алифатическими диазосоединениями. Стабильные алифатические диазосоединения, например производные диазоацетамида, в присутствии ионов меди реагируют с некоторыми карбоксильными группами, например с карбоксильной группой одного из остатков аспарагиновой кислоты в активном центре пепсина или других аспартильных протеиназ:
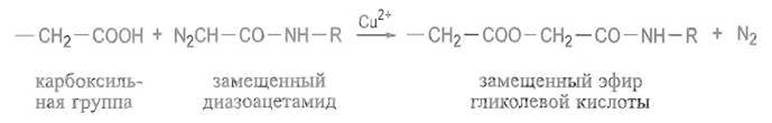
Алкилирование карбоксильных групп. В качестве агентов, алкилирующих некоторые карбоксильные группы в белках, могут использоваться а-галоидкетоны, например n-бромфенацилбромид Br—СН2СС—С6Н4Вr и его аналоги, а также а-галоидкислоты. Реакция с n-бромфенацилбромидом протекает следующим образом:

9.2.4. Модификация остатков метионина
Остатки метионина в белках и особенно пептидах легко окисляются до метионинсулъфоксида:
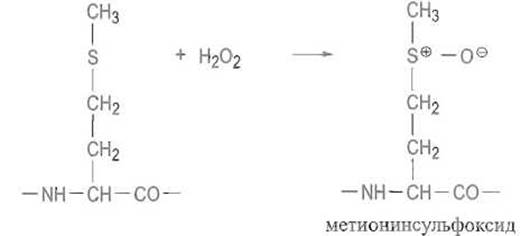
В ряде случаев эта реакция протекает даже под действием кислорода воздуха. В более жестких условиях, например при действии надмуравьиной кислоты, окисление проходит глубже и приводит к метионинсульфону:
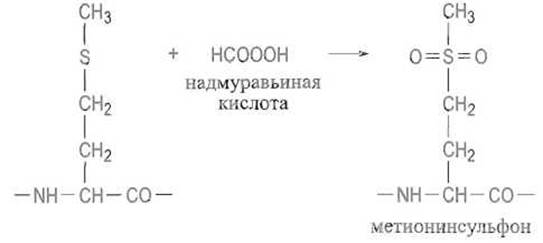
Для модифицирования остатков метионина чаще, однако, применяют алкилирующие агенты, например ужe упоминавшийся n-бромфенацилбромид или а-галоидзамещенные кислоты, в частности иодуксусную кислоту или ее амид. При этом характерно, что сера метионина реагирует с такого рода соединениями даже в слабокислой среде, когда большинство других нуклеофильных групп белка, в частности имидазольные группы гистидина, в реакцию не вступают, будучи протонированы:

Аналогично протекает реакция остатков метионина с а-бромкетонами.
9.2.5. Модификация остатков цистеина
Сульфгидрильные группы остатков цистеина весьма реакционноспособны и могут как участвовать как в реакциях замещения в качестве нуклеофильного компонента, чему благоприятствует их диссоциация с образованием тиолят-иона (рКа около 8), так и подвергаться окислению или образовывать прочные соли с ионами тяжелых металлов.
Алкилирование тиоловых групп. Тиоловые группы легко алкилируются в слабощелочной среде. Например, при действии иод- уксусной кислоты остатки цистеина превращаются в S-карбоксиметилцистеин, при действии иодацетамида — в S-карбоксамидометилцистеин:
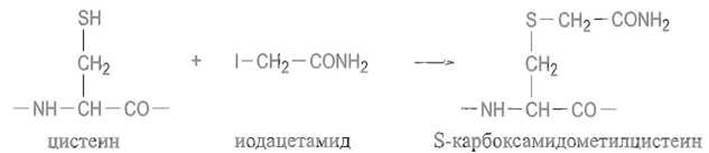
Тиоловая группа цистеина легко присоединяется к двойным связям, если они активированы сопряженной карбонильной группой или другим способом:
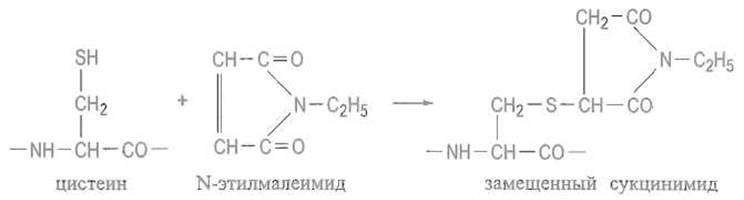
Окисление сульфгидрильных групп. Сульфгидрильные группы остатков цистеина могут окисляться с образованием дисудьфидных групп, т.е. превращаться в остатки цистина в сравнительно мягких условиях, в частности под действием кислорода воздуха. Эта реакция специфична и может сопровождаться лишь окислением серы в метионине. Иногда, в частности при ренатурации белков, для перевода остатков цистеина в остатки цистина прибегают к реакции дисульфидного обмена, для чего особенно подходящей считается окисленная форма глутатиона:
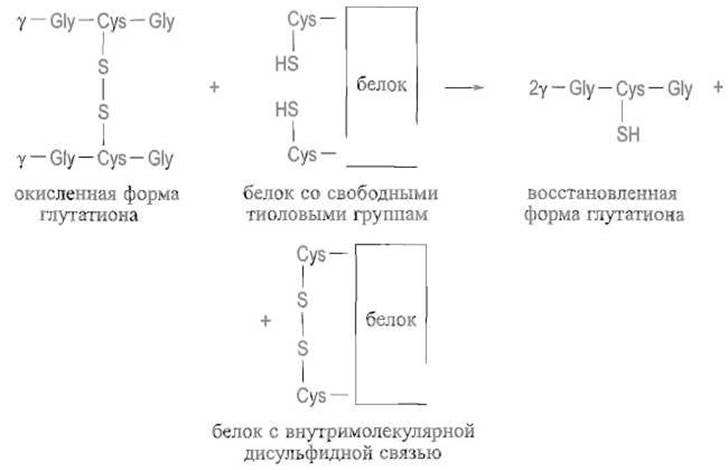
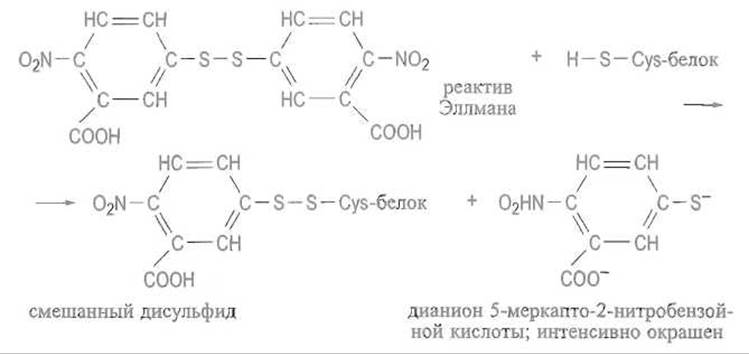
Разумеется, эта реакция обратима, однако равновесие может быть практически полностью сдвинуто в сторону образования дисульфидной (как правило, внутримолекулярной) связи в белке за счет большого избытка окисленной формы глутатиона. На реакции дисульфидного обмена основано использование так называемого реактива Эллмана — 5,5'-дитиобие-(2-нитробензойной) кислоты для количественного определения свободных сульфгидрильных групп в белках. При взаимодействии с последними происходит восстановление дисульфидной связи в реактиве. Образующаяся 5-меркапто-2-нитробензойная кислота за счет диссоциации сульфгидрильной группы в щелочной среде дает характерное желтое окрашивание и может быть количественно определена спектрофотометрически:
Образование меркаптидов металлов. Сульфгидрильная группа, будучи ионизована, т.е. в форме меркаптид-иона, специфически взаимодействует с катионами многих тяжелых металлов и металлоорганическими соединениями, образуя весьма прочные меркаптиды. Аналитическое значение имеют реакции с катионом двухвалентной ртути:
![]()
а также n-гидрокси- (или n-хлор-)меркурбензоатом

Связь Hg—S в таких соединениях занимает промежуточное положение между ионной и ковалентной и, как правило, достаточно прочна. Образование меркаптида n-меркурбензойной кислоты сопровождается характерным изменением УФ-спектра, что может быть использовано для количественного определения свободных сульфгидрильных групп в белках и пептидах, хотя этот способ менее удобен, чем реакция Эллмана.
9.2.6. Модификация имидазольной группы гистидина
Алкилирование. Имидазол гистидина легко вступает в реакции алкилирования, например с иодуксусной кислотой или ее амидом в нейтральной или слабощелочной среде, причем в зависимости от микроокружения этой группы в белке или способа связывания алкилирующего реагента реакция может происходить по одному или по другому атому азота (по N—1 или N—3):
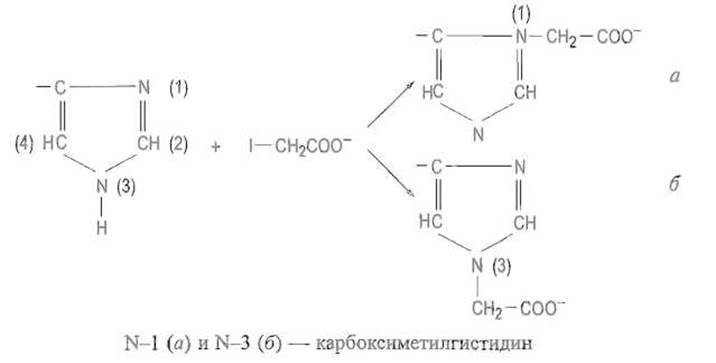
Следует учитывать, что двойная связь в системе N=C—N имидазола гистидина способна перемещаться и занимать любое из двух положений. Реакция алкилирования не является характерной только для гистидина, она может протекать также с остатками метионина, лизина и даже с некоторыми карбоксильными группами, что зависит от pH и в значительно большей мере от особенностей микроокружения модифицируемого остатка.
Азосочетание. Реакция азосочетания может затрагивать в имидазольной группе гистидина как атом С-2, так и атом С-4:
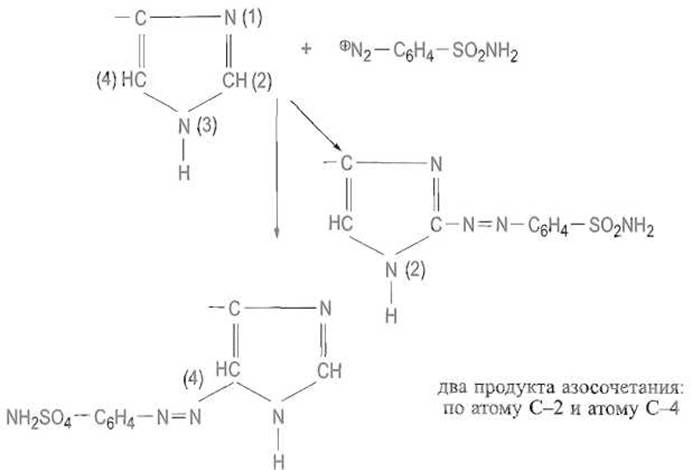
Продукты азосочетания гистидина с различными солями диазония (на схеме показана реакция с амидом диазосульфаниловой кислоты) имеют интенсивную, обычно красно-оранжевую окраску (реакция Паули).
Для избирательной деструкции остатков гистидина прибегают к фотоокислению кислородом в присутствии сенсибилизирующих красителей.
9.2.7. Модификация гуанидогруппы аргинина
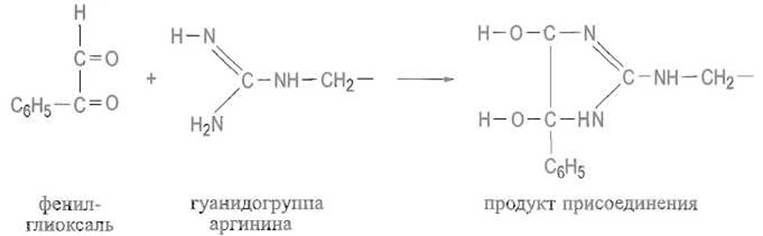
Эта задача является особенно трудной, поскольку δ-гуанидогруппа аргинина является очень сильным основанием (рКа≈ 14) и может быть депротонирована, что позволило бы ей выступать в качестве нуклеофила лишь в крайне жестких условиях, при такой концентрации щелочи, которая обрекает белок на глубокую денатурацию. Удается, однако, использовать для избирательной модификации аргинина способность гуанидогруппы вступать в реакции образования гетероциклов с некоторыми дикетонами или апьдегидокетонами:
9.2.8. Модификация индола в триптофане
Индольная группировка весьма лабильна, в частности, она претерпевает деструкцию при фотосенсибилизировакном окислении. Среди химических методов упомянем реакцию с о-нитрофенилсульфенилхлоридом, которая протекает по а-углеродному атому пиррольной части индола:

Заканчивая краткий обзор реагентов, используемых при модификации отдельных аминокислотных остатков в белках, отметим, что ко многим аминокислотам этот метод вовсе не применим Таковы все гидрофобные аминокислоты, глицин, пролин. Нет общих подходов к модификации серина, треонина, аспарагина и глутамина.