ОСНОВЫ БИОХИМИИ ЛЕНИНДЖЕРА - ТОМ 1. ОСНОВЫ БИОХИМИИ СТРОЕНИЕ И КАТАЛИЗ - 2011
ЧАСТЬ I. СТРОЕНИЕ И КАТАЛИЗ
6. ФЕРМЕНТЫ
6.4. Примеры ферментативных реакций
До сих пор мы говорили об общих принципах катализа и ввели некоторые кинетические параметры для описания действия ферментов. Теперь перейдем к рассмотрению отдельных примеров механизмов ферментативных реакций.
Для полного понимания механизма действия выделенного фермента необходимо идентифицировать все его субстраты, кофакторы, продукты и регуляторные молекулы. Более того, необходимо узнать 1) порядок стадий ферментативной реакции, 2) структуру каждого промежуточного соединения (интермедиата), 3) скорости взаимных превращений интермедиатов, 4) структурные взаимоотношения фермента с каждым интермедиатом, 5) энергетический вклад каждой связи, образующейся в промежуточных комплексах и переходных состояниях. На сегодняшний день, вероятно, нет ни одного фермента, для которого ответы на все эти вопросы были бы получены. Однако десятки лет исследовательской работы привели к накоплению большого количества информации, в некоторых случаях весьма подробной, о механизмах действия сотен ферментов.
Далее мы остановимся на механизмах действия четырех ферментов: химотрипсипа, гексокиназы, енолазы и лизоцима. Эти примеры, безусловно, не могут отразить все многообразие ферментативных реакций. Подобный выбор объясняется отчасти тем, что эти ферменты относятся к разряду наиболее изученных, кроме того, на их примере удобно проиллюстрировать некоторые общие принципы, описанные в данной главе. При обсуждении принципов действия ферментов мы будем говорить и о ключевых экспериментах, которые позволили их установить. На примере химотрипсина мы еще раз остановимся на тех правилах, которые используются для описания механизмов ферментативных реакций. Многие детали механизмов и экспериментальных данных, конечно, приходится опускать, поскольку ни одна книга не смогла бы вместить в себя всю богатейшую историю экспериментов по изучению этих ферментов. Кроме того, здесь мы не будем специально останавливаться на роли коферментов в ферментативной реакции. Различным функциям коферментов больше внимания будет уделено в части II.
Механизм действия химотрипсина включает стадии ацилирования и деацилирования остатка серина
Химотрипсин из поджелудочной железы быка (Мr = 25 191) является протеазой — ферментом, катализирующим расщепление пептидной связи. Данная протеаза специфическим образом расщепляет пептидные связи, которыми соединены остатки ароматических аминокислот (Тrр, Рhе и Туr). На рис. 6-18 представлена трехмерная структура химотрипсина и отмечены функциональные группы в его активном центре. Реакция, катализируемая данным ферментом, иллюстрирует принцип стабилизации переходного состояния, а также является классическим примером общего кислотно-основного и ковалентного катализа.
Рис. 6-18. Структура химотрипсина (PDB ID 7GCH). а) Первичная структура белка, на которой отмечены дисульфидные связи и важные для катализа аминокислотные остатки. Белок образован тремя полипептидными цепями, соединенными дисульфидными мостиками (нумерация остатков химотрипсина с «отсутствующими» остатками 14, 15, 147 и 148 объясняется на рис. 6-38). В трехмерной структуре белка аминокислотные остатки, образующие активный центр, сгруппированы вместе. 6) Изображение поверхности белка. Зеленым цветом выделен карман, в котором происходит связывание ароматической боковой цепи аминокислотного остатка субстрата. Красным цветом выделены ключевые остатки активного центра: Ser195, His57 и Asp102. Роль этих остатков в катализе отражена на рис. 6-21. в) Полипептидный остов белка изображен в виде ленты. Дисулъфидные связи выделены желтым цветом, а три полипептидные цепи изображены теми же цветами, что и на рисунке (а), г) Закрытие активного центра при связывании субстрата (основная часть изображена зеленым цветом). Частично видны два остатка активного центра — Ser195и His57 (красные). Гидроксильная группа Ser195 атакует карбонильную группу субстрата (кислород карбонильной группы выделен сиреневым цветом); образующийся на кислороде отрицательный заряд стабилизируется в полости, образованной амидными атомами азота (в том числе от Ser195, показанного оранжевым цветом), как объясняется на рис. 6-21. Синим цветом изображены боковая цепь ароматического аминокислотного остатка и амидный азот расщепляемой пептидной связи (направлены в сторону к читателю и закрывают собой остальную полипептидную цепь субстрата).

Химотрипсин ускоряет гидролиз пептидной связи как минимум в 109 раз. Он не катализирует прямую атаку пептидной связи молекулой воды, а участвует в образовании переходного ацилферментного комплекса. Данная реакция имеет две четко различимые стадии. На стадии ацилирования происходит расщепление пептидной связи и образование сложноэфирной связи между карбонильной группой пептида и ферментом. На стадии деацилирования происходит гидролиз сложноэфирной связи и высвобождение исходной формы фермента.
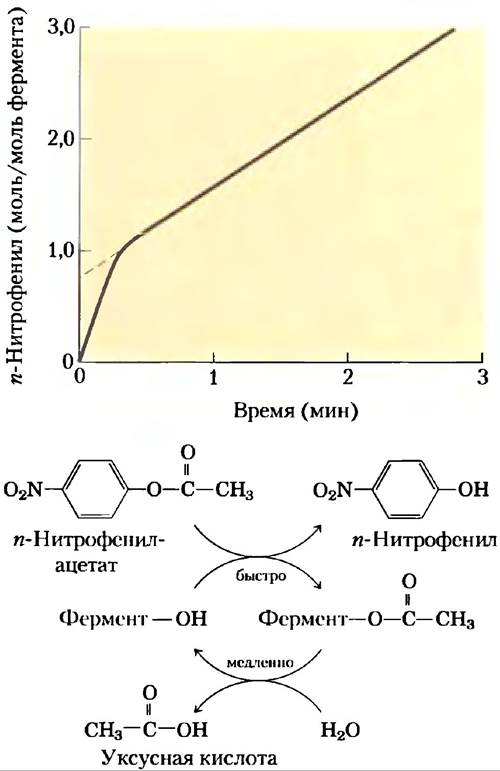
Наличие ковалентного ацилферментного производного было продемонстрировано методами предстационарной кинетики. Химотрипсин гидролизует не только пептиды, но и небольшие молекулы сложных эфиров и амидов. Данные реакции протекают гораздо медленнее, чем гидролиз пептидов, поскольку энергия связывания с небольшими субстратами гораздо ниже; в результате такие реакции легче изучать. В 1954 г. Б. С. Хартли и Б. А. Килби обнаружили, что скорость гидролиза n-нитрофенилацетата под действием химотрипсина, определявшаяся по образованию n-нитрофенола, сначала была очень высокой, а потом снижалась (рис. 6-19). Проведя экстраполяцию к нулевому моменту времени, они заключили, что быстрая фаза соответствует ситуации, при которой на каждую молекулу фермента приходится образование одной молекулы n-нитрофенола. Хартли и Килби предположили, что в данной реакции происходит быстрое ацилирование всего имеющегося фермента (и высвобождение n-нитрофенола) с последующим медленным его деацилированием. Аналогичные результаты были получены для многих других ферментов. Возможность обнаружения быстрой стадии реакции является еще одним примером использования кинетических методов для идентификации реакционных стадий.
Рис. 6-19. Применение предстационарной кинетики для доказательства образования ацилферментного производного. За гидролизом n-нитрофенилацетата следили по образованию окрашенного продукта n-нитрофенола. В начале скорость образования n-нитрофенола находится почти в стехиометрическом соответствии с количеством фермента — это быстрая стадия ацилирования. Затем скорость снижается, поскольку высвобождение фермента лимитирует медленная реакция деацилирования.
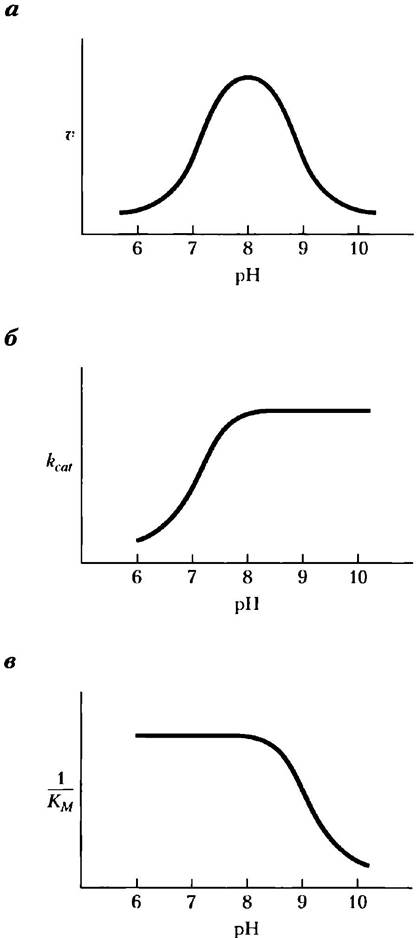
Дополнительные данные о механизме действия химотрипсина были получены при анализе влияния pH на скорость реакции. Зависимость скорости гидролитического расщепления субстратов под действием химотрипсина от pH обычно имеет колоколообразный вид (рис. 6-20). Скорости реакции на рис. 6-20, а были определены принизкой (ненасыщающей) концентрации субстрата и, следовательно, отражают отношение rcat /Км. Данный график можно разделить на составляющие части. Для этого следует определить максимальную скорость при каждом значении pH и изобразить зависимость rcat от pH (рис. 6-20, б). После определения Км для каждого значения pH можно также построить график зависимости 1 /Км от pH (рис. 6-20, в). Анализ данных кинетики и структуры показал, что изменение rcat связано с состоянием ионизации His57. Снижение величины rcat при уменьшении pH происходит из-за протонирования остатка His57, который более не способен забирать протон от Ser195на первой стадии реакции (рис. 6-21). Такое снижение скорости указывает на важную роль общего основного и общего кислотного катализа в механизме действия химотрипсина. Изменения параметра 1 /Кмотражают ионизацию α-аминогруппы остатка Ilе16, расположенного на N-конце одной из трех полипептидных цепей химотрипсина. Эта группа участвует в образовании солевого мостика с Asp194, что стабилизирует активную конформацию фермента. Когда при повышении pH эта группа теряет протон, солевой мостик распадается, и происходящие конформационные изменения закрывают гидрофобный карман, куда помещается боковая цепь ароматического аминокислотного остатка субстрата (рис. 6-18). В результате субстрат не может полноценно связываться, что выражается в увеличении константы Михаэлиса.
Рис. 6-20. Зависимость активности химотрипсина от pH. а) Зависимость скорости гидролитического расщепления субстратов под действием химотрипсина от pH имеет колоколообразный вид с максимумом при pH 8,0. Скорость v была рассчитана при низкой концентрации субстрата и поэтому отражает отношение rcat/KM. С помощью кинетических методов можно определить раздельно значения rcat и КM и построить график зависимости каждого из этих параметров от pH (б и в). Из этих графиков видно, что переход при pH> 7 связан с изменением rcat, а при pH> 8,5 — с изменением Км. Результаты кинетических и структурных исследований показали, что эти переходы отражают изменения состояния ионизации соответственно боковой цепи His57 (при отсутствии связывания субстрата) и α-амино- группы Ilе16 (на N-конце цепи В). Для оптимальной активности фермента His57 должен находиться в непротонированном состояния, а Ilе16 должен быть протонирован.
Нуклеофилом на стадии ацилирования выступает кислород остатка Ser193. (Протеазы, в каталитическом механизме действия которых принимает участие остаток Ser, называют сериновыми протеазами.) Значение рКa гидроксильной группы Ser обычно слишком высокое, чтобы непротонированная форма могла встречаться в значительной концентрации при физиологических значениях pH. Однако в молекуле химотрипсина Ser195связан водородными связями с His57 и Asp102 в так называемую каталитическую триаду. При связывании пептидного субстрата с химотрипсином происходит небольшое изменение конформации белка, укорачивающее водородную связь между His57 и Asp102, что приводит к более сильному взаимодействию (сильная водородная связь). Это в свою очередь повышает рКаHis57 от -7 (в свободном гистидине) до >12, что позволяет гистидину действовать в качестве более сильного общего основания и удалять протон из гидроксильной группы Ser193. Удаление протона предотвращает образование очень неустойчивого положительного заряда па гидроксильной группе Ser195и делает боковую цепь Ser сильным нуклеофилом. На более поздних стадиях реакции His57 выступает в качестве донора протона и протонирует аминогруппу в уходящей группе (в удаляемой части субстрата).
При атаке карбонильной группы субстрата кислородом остатка Ser195образуется короткоживущее четвертичное промежуточное соединение, в котором карбонильный кислород имеет отрицательный заряд (рис. 6-21). Этот заряд, локализованный в кармане молекулы фермента, называемом оксианионовой дырой, стабилизируется водородными связями с участием амидных групп двух пептидных связей в молекуле химотрипсина. Одна из этих водородных связей (с участием Gly193) присутствует только в данном промежуточном соединении, а также в переходных состояниях при его образовании и распаде; она снижает уровень энергии, необходимый для достижения данных состояний. Это один из примеров, иллюстрирующих роль энергии связывания в катализе.
Значение комплементарности фермента переходному состоянию для осуществления ферментативного катализа обсуждается в доп. 6-3.
Учимся определять механизм реакции
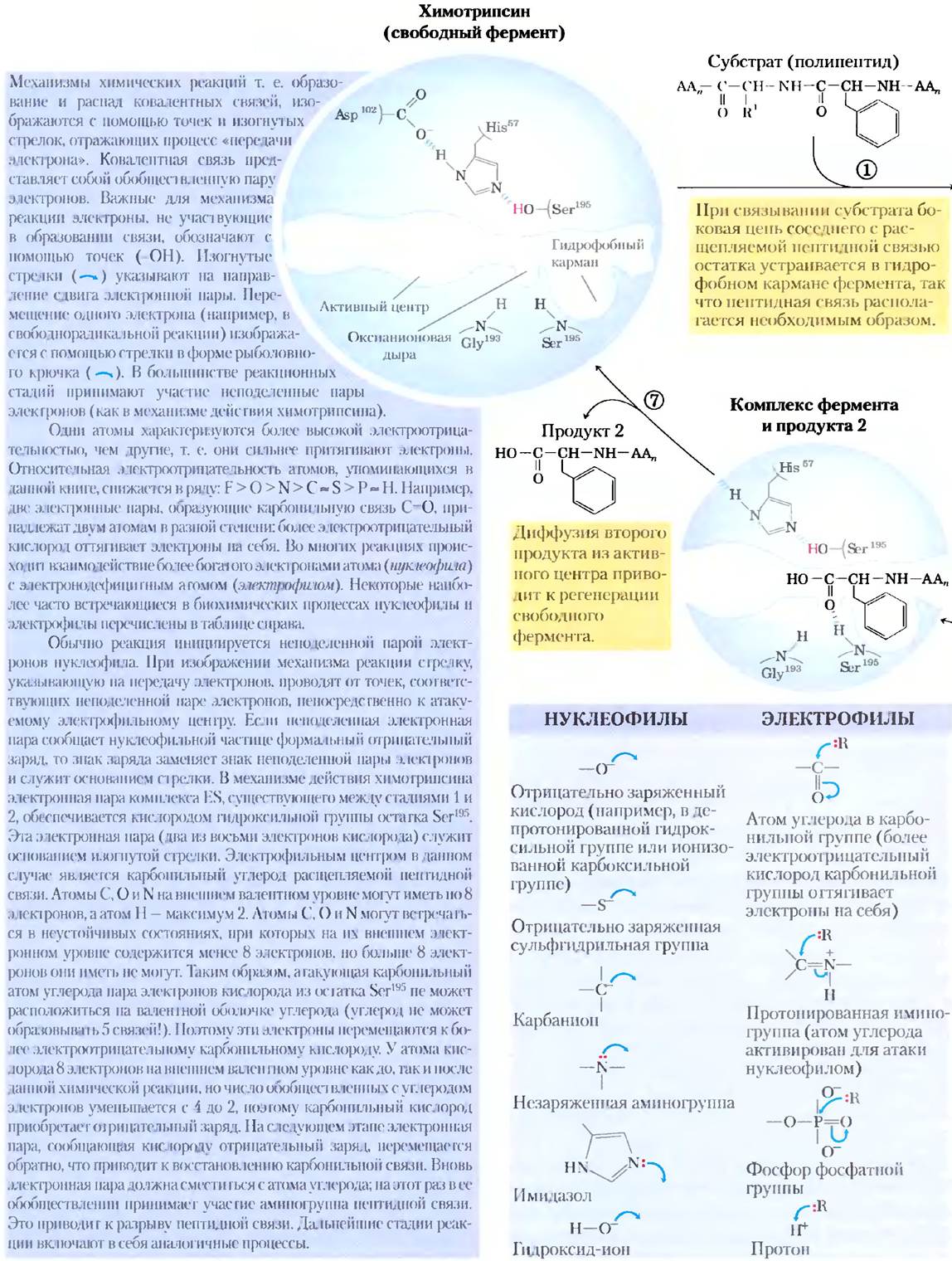
Рис. 6-21. Механизм гидролитического расщепления пептидной связи под действием химотрипсина. Реакция протекает в два этапа. На первом этапе (стадии 1-3) происходит образование ковалентного ацилферментного комплекса, сопряженное с расщеплением пептидной связи. На стадии деацилирования (стадии 4-7) происходит регенерация фермента. Данный этап в основном представляет собой процесс, обратный ацилированию, в котором вода исполняет роль аминокомпонента субстрата.

* Четвертичные промежуточные соединения, образующиеся в катализируемой химотрипсином реакции, иногда называют переходным состоянием, что может внести путаницу. Промежуточное соединение (интермедиат) представляет собой химическое соединение с ограниченным временем жизни, превышающим время одного молекулярного колебания (≈10-13 с). Переходное состояние — это участок координаты реакции с максимальной свободной энергией; оно не характеризуется временем жизни. Четвертичные интермедиаты, образующиеся на двух этапах данной реакции, очень похожи как по энергетическому состоянию, так и по структуре; переходные состояния возникают при образовании и распаде этих интермедиатов. Таким образом, интермедиат является обязательной стадией образования связи, а переходное состояние — это часть реакционного процесса. В случае химотрипсина тесная связь между интермедиатом и переходными состояниями иногда приводит к смешению этих понятий. Более того, взаимодействие отрицательно заряженного кислорода с амидными атомами азота в оксианионовой дыре, которое часто называют стабилизацией переходного состояния, в этом случае играет роль и в стабилизации интермедиата. Не все промежуточные соединения являются настолько короткоживущими, что напоминают переходные состояния. Ацилферментное производное химотрипсина — это гораздо более устойчивое промежуточное соединение, которое можно легко детектировать и изучать; его никогда не путают с переходным состоянием.
Доказательства комплементарности фермента переходному состоянию
Переходное состояние реакции трудно изучать, поскольку оно слишком короткоживущее. Однако для понимания механизма катализа необходимо анализировать взаимодействие между ферментом и этим мимолетным состоянием субстрата в ходе реакции. Комплементарность фермента и переходного состояния можно считать обязательным условием катализа, поскольку изменение энергетического барьера, соответствующего переходному состоянию, происходит только в том случае, если имеет место катализ. Как получить доказательства комплементарности фермента и переходного состояния? К счастью, в руках исследователей есть множество способов — как старых, так и новых — для решения этой проблемы. Каждый из этих способов вносит свой вклад в обоснование основного принципа действия ферментов.
Связь структуры и активности
Если фермент комплементарен переходному состоянию реакции, то некоторые функциональные группы как фермента, так и субстрата должны взаимодействовать преимущественно не в ЕS-комплсксе, а в переходном состоянии. Изменение этих групп не должно сильно сказываться на образовании комплекса и, следовательно, не должно заметно изменить такие константы, как константа диссоциации (Кd; или Км, если Кd = Км), характеризующие равновесие Е + S ⇄ ЕS. Но изменение тех же групп должно сильно отразиться на общей скорости реакции (т. е. на значении rcat или отношении rcat / Км), поскольку при связывании субстрата нс происходит тех взаимодействий, которые могли бы снизить энергию активации.
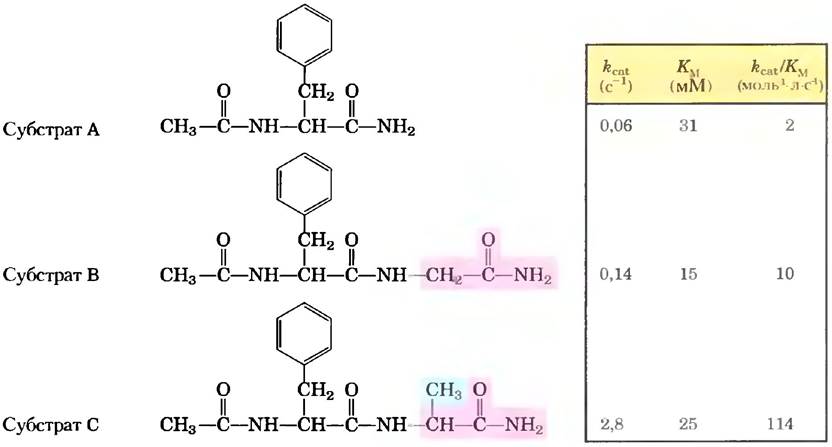
Превосходной иллюстрацией данного эффекта являются кинетические эксперименты с серией родственных субстратов химотрипсина (рис. 1). В норме химотрипсин гидролизует пептидную связь, расположенную рядом с ароматической аминокислотой. На рис. 1 приведены формулы низкомолекулярных субстратов, использовавшиеся в качестве моделей природных субстратов — длинных пептидов или белков. Добавляемая в каждом следующем субстрате (от А через В к С) химическая группа выделена цветом. Как видно из таблицы, добавление групп к молекуле субстрата почти не оказывало влияния на константы Михаэлиса, однако сильно увеличивало rcat и rcar/ Kм. Именно этого эффекта и следует ожидать в том случае, если взаимодействие между ферментом и субстратом больше влияет на стабилизацию переходного состояния. Данные результаты также показывают, что скорость реакции в значительной степени зависит от фермент-субстратных взаимодействий, происходящих в участках, удаленных от реагирующих групп. Более подробно механизм действия химотринсина описан в основном тексте.
Рис. 1. Влияние небольших изменений структуры субстрата на кинетические параметры гидролиза амидов под действием химотрипсина.
Дополнительным методом анализа может служить модификация самого фермента, приводящая к ликвидации некоторых фермент-субстратных взаимодействий. Этого можно добиться, вызывая замены определенных аминокислотных остатков путем направленного мутагенеза (рис. 9-11). Результаты подобных экспериментов также демонстрируют важность энергии связывания для стабилизации переходного состояния.
Аналоги переходного состояния
Даже если переходное состояние нельзя увидеть непосредственно, химик часто может предсказать его примерную структуру, основываясь на данных о механизме реакции. Переходное состояние по определению является временным и настолько неустойчивым, что прямые измерения взаимодействий между интермедиатом и ферментом не представляются возможными. Однако в некоторых случаях удается создать устойчивые молекулы, структура которых напоминает переходное состояние. Их называют аналогами переходного состояния. В принципе, эти вещества должны связываться с ферментом более прочно, чем связан субстрат в комплексе ЕS, поскольку они лучше соответствуют структуре активного центра (т. е. образуют большее количество слабых связей), чем сам субстрат. Идея создания аналогов переходного состояния была высказана Полингом в 1940-х гг. и воплощена в жизнь для нескольких ферментов. Ограничением данного подхода является то, что аналоги не могут абсолютно точно имитировать переходное состояние. Некоторые из них связываются с ферментом в 102-106раз прочнее, чем обычные субстраты, что служит доказательством комплементарности активного центра фермента именно переходному состоянию. Тот же принцип используется сегодня в фармацевтической промышленности при создании новых лекарственных препаратов. Мощные лекарства, направленные на борьбу с ВИЧ- инфекцией, — ингибиторы протеаз — создавались как прочно связывающиеся аналоги переходного состояния для связывания с активным центром протеазы ВИЧ.
Каталитические антитела
Если можно создать аналог переходного состояния для реакции S ⇄ Р, то антитела, прочно связывающиеся с этими аналогами, возможно, окажутся способнымикатализировать реакцию S —> Р. Антитела (или иммуноглобулины, рис. 5-23) играют ключевую роль в действии иммунной системы. Если аналог переходного состояния использовать в качестве эпитопа для стимуляции продукции антител, то связывающиеся с ним антитела являются потенциальными катализаторами соответствующей реакции. Использование таких «каталитических антител» (их часто называют абзимами от англ. antibody и enzyme. — Прим. ред.) впервые было предложено В. П. Дженксом в 1969 г. и вошло в практику с развитием лабораторных методов, позволяющих получать достаточные количества идентичных антител, связывающихся с одним специфическим антигеном (моноклональных антител, гл. 5).
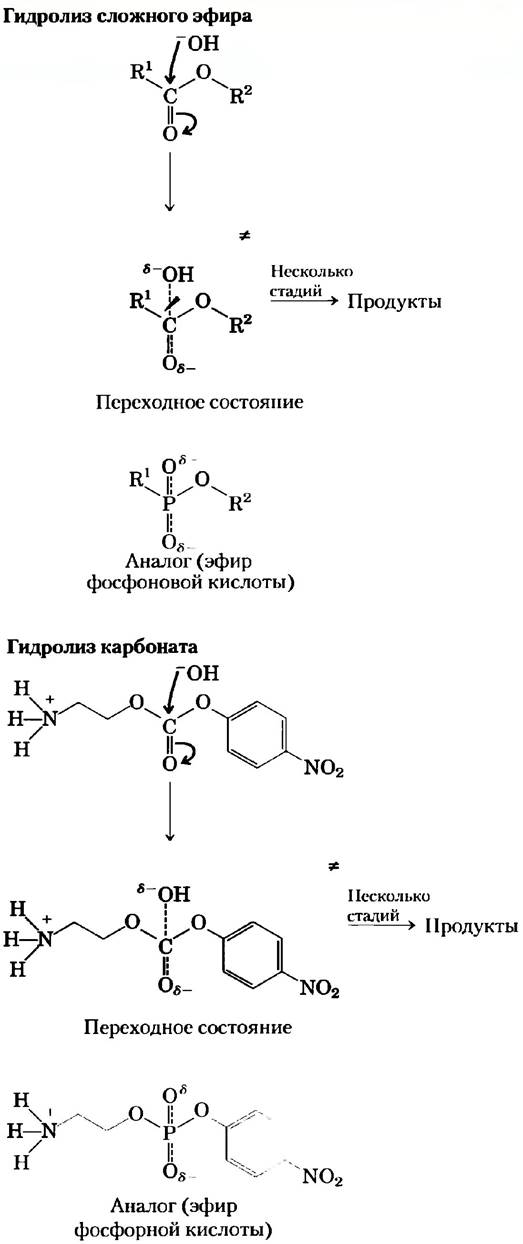
Пионерные работы, выполненные в лабораториях Ричарда Лернера и Петера Шульца, привели к выделению ряда моноклональных антител, катализирующих гидролиз сложных эфиров или карбонатов (рис. 2). В этих реакциях в результате атаки воды (ОII) на карбонильный углерод образуется четвертичное переходное состояние, в котором на карбонильном атоме кислорода сосредоточен частичный отрицательный заряд. Эфиры фосфоповой кислоты похожи по структуре и заряду на переходное состояние в реакции гидролиза сложных эфиров, что делает возможным их использование в качестве аналогов переходного состояния. Для гидролиза карбонатов используются эфиры фосфорной кислоты. Было обнаружено, что прочно связывающиеся с фосфонатами или фосфатами антитела ускоряют соответствующие реакции гидролиза сложного эфира или карбоната в 103-104 раз. Структурный анализ нескольких из этих каталитических антител показал, что боковые цепи определенных аминокислотных остатков расположены таким образом, что они могли бы взаимодействовать с субстратом в переходном состоянии.
Рис. 2. Предполагаемые переходные состояния для реакций гидролиза сложных эфиров и карбонатов. Хорошими аналогами переходного состояния для этих реакций являются эфиры фосфоновой и фосфорной кислоты соответственно.
Каталитические антитела обычно не достигают каталитической эффективности ферментов, однако возможность их медицинского и промышленного применения очевидна. Например, было предложено использовать каталитические антитела, созданные для деградации кокаина, при лечении кокаиновой зависимости.
Индуцированное соответствие при связывании субстрата с гексокиназой
Гексокиназа дрожжей (Мr = 107 862) катализирует обратимую двухсубстратную реакцию:

АТР и ADP всегда связываются с ферментами в виде комплексов с ионами Mg2+.
Гидроксильная группа у атома С-6 глюкозы (на которую в процессе ферментативной реакции переносится y-фосфорильная группа АТР) по реакционной способности близка к воде; кроме того, вода легко проникает в активный центр фермента. Однако присутствие гексокиназы в 106 раз повышает вероятность протекания реакции с глюкозой. Способность фермента различать воду и глюкозу связана с конформационными изменениями в его молекуле, происходящими при связывании «правильного» субстрата (рис. 6-22). Таким образом, механизм действия гексокиназы является хорошим примером индуцированного соответствия. Если в среде нет глюкозы, то фермент пребывает в неактивном состоянии, когда аминокислотные остатки активного центра находятся в нереакционноспособном положении. При связывании глюкозы (но не воды) и Mg • АТР образующаяся энергия связывания приводит к конформационным изменениям в молекуле гексокиназы, переводящим ее в активное состояние.
Рис. 6-22. Индуцированное соответствие в молекуле гексокиназы. а) Гексокиназа имеет U-образную форму (РDВ ID 2YНХ). б) Связывание D-глюкозы (выделена красным цветом) приводит к конформационным изменениям, сопровождающимся соединением двух концов молекулы (РDВ ID 1HKG и РDВ ID 1GLК).

Данная модель была подтверждена кинетическими исследованиями. Пятиуглеродный сахар ксилоза, по стереохимическим свойствам напоминающий глюкозу, но содержащий на один атом углерода меньше, связывается с гексокиназой, но не может фосфорилироваться. Тем не менее добавление в реакционную смесь ксилозы увеличивает скорость гидролиза АТР. Очевидно, связывания ксилозы достаточно, чтобы вызвать переход гексокиназы в активное конформационное состояние, и «обманутый» фермент фосфорилирует воду. Эта реакция гексокиназы также демонстрирует, что специфичность фермента не всегда заключается в связывании одного-единственного вещества. В данном случае специфичность фермента не в образовании комплекса ЕS, а в отношении скоростей последующих каталитических стадий. Для проникновения воды в активный центр нет препятствий, но скорость реакции сильно возрастает в присутствии функционального акцептора фосфорильной группы (глюкозы).

Индуцированное соответствие является лишь одним аспектом механизма действия гексокиназы. Подобно химотрипсину, этот фермент также использует несколько стратегий катализа. Например, аминокислотные остатки активного центра (те, что принимают правильное положение в результате конформационных изменений при связывании субстрата) участвуют в общем кислотно-основном катализе и стабилизации переходного состояния.
Механизм реакции енолазы требует присутствия ионов металла
Еще один гликолитический фермент — енолаза — катализирует обратимую дегидратацию 2-фосфоглицсрата с образованием фосфоенолпирувата:
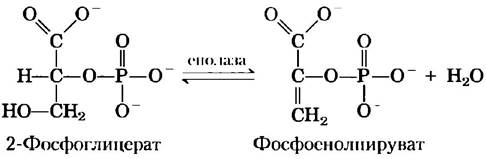
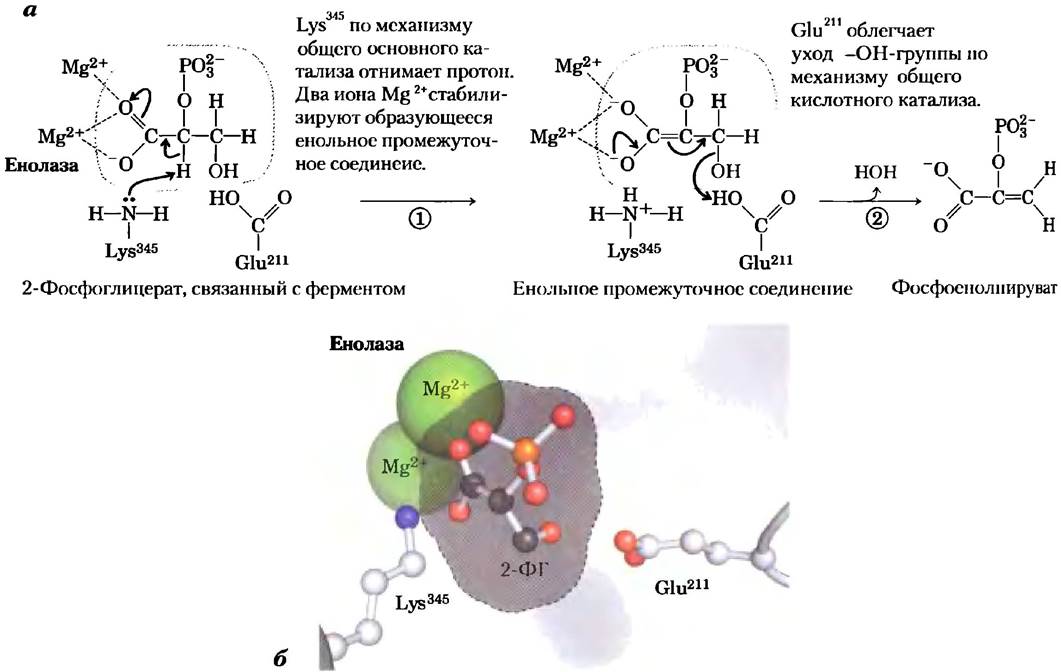
Енолаза дрожжей (Мr = 93 316) представляет собой димер, каждая субъединица которого состоит из 436 аминокислотных остатков. Ферментативная реакция с участием енолазы иллюстрирует один из типов катализа с участием ионов металлов, а также является еще одним примером общего кислотно-основного катализа и стабилизации переходного состояния. Реакция протекает в две стадии (рис. 6-23, а). Сначала остаток Lys345 действует в качестве общего основания, отнимая протон от атома С-2-2-фосфоглицерата. Затем остаток Glu211выступает в роли общей кислоты и отдает протон уходящей ОН-группе. Протон в С-2 положении 2-фосфоглицерата не очень «кислый» и поэтому не слишком легко удаляется. Однако в активном центре фермента 2-фосфоглицерат участвует в довольно сильных ионных взаимодействиях с двумя связанными ионами Mg2- (рис. 6-23, б). В результате протон у атома С-2 становится более «кислым» (снижается значение рКa) и удаляется легче. В каталитическом механизме задействованы также водородные связи с участием других аминокислотных остатков активного центра. Различные взаимодействия приводят к эффективной стабилизации как енольного промежуточного соединения, так и переходного состояния, предшествующего его образованию.
Рис. 6-23. Механизм реакции. Двухстадийная реакция, катализируемая енолазой. а) Механизм превращения 2-фосфоглицерата (2-ФГ) в фосфоенолпируват. Карбоксильная группа 2-ФГ координирована двумя ионами магния в активном центре фермента, б) Положение субстрата, ионов Mg2+, Lys 345и Glu211в активном центре енолазы. Атомы водорода не показаны. Все атомы кислорода в молекуле 2-ФГ изображены розовым цветом, а атом фосфора — оранжевым (PDB ID 1ONE).
Механизм действия лизоцима включает две последовательные стадии нуклеофильного замещения
Лизоцим — это природное антибактериальное вещество, присутствующее в слезах и яичном белке. Лизоцим куриного белка (Мr = 14 296) представляет собой мономер, состоящий из 129 аминокислотных остатков. Он был первым ферментом, для которого в 1965 г. Дэвид Филлипс с сотрудниками определили трехмерную структуру. Молекула лизоцима удерживается четырьмя дисульфидными мостиками и имеет длинную расщелину, в которой расположен его активный центр (рис. 6-24, а). Более пяти десятилетий активного изучения лизоцима привели к созданию детальной картины, отражающей его структуру и активность; эти исследования являются показателем развития биохимической науки.
Рис. 6-24. Структура лизоцима яичного белка и катализируемая им реакция. а) Ленточная модель структуры белка: остатки Glu35и Asp52 в активном центре фермента изображены в виде синих стержней, а связанный субстрат выделен красным цветом (PDB ID 1LZE). б) Реакция, катализируемая лизоцимом яичного белка. Изображен фрагмент пептидогликана с указанием участков связывания в молекуле фермента, обозначенных буквами от А до F. Красная стрелка указывает на расщепляемую гликозидную связь С-О, расположенную между остатками сахаров, локализованных в участках связывания D и Е. Во вставке изображена сама реакция; красным цветом выделен кислород, происходящий из молекулы воды. Принятые сокращения: Mur2Ac — N-ацетилмурамовая кислота, GlcNAc — N-ацетилглюкозамин, RО — остаток молочной кислоты, NAc и AcN — N-ацетильная группа.

Субстратами лизоцима служат пептидогликаны - углеводы, обнаруженные в клеточных стенках многих бактерий (см. рис. 20-31). Лизоцим расщепляет β1 —> 4 гликозидную С—О связь (см.с. 348) между двумя типами остатков сахара в молекуле: остатками N-ацетилмурамовой кислоты (Mur2Ac) и N-ацетилглюкозамина (GlcNAc), которые часто обозначают соответственно, как NAM и NAG (рис. 6-24, б). Шесть чередующихся остатков этих сахаров в молекуле пептидогликана связываются
в активном центре фермента в позициях, обозначаемых буквами от А до F. Исследования на модальных структурах показали, что боковая цепь N-ацетилмурамовой кислоты не позволяет этому остатку связываться в позициях С и Е, а только в В, D или F. При связывании пептидогликапа расщепляется лишь одна гликозидная связь, соединяющая остаток N-ацетилмурамовой кислоты в положении D и остаток N-ацетилглюкозамина в положении Е. Ключевыми аминокислотными остатками фермента являются Glu35и Asp52(рис. 6-25, а). Реакция идет по механизму нуклеофильного замещения: ОН-группа воды замещает остаток N-ацетилглюкозамина у С-1 атома N-ацетилмурамовой кислоты.
Рис. 6-25. Механизм реакции. Действие лизоцима. В результате данной реакции вода, присоединяющаяся к атому С-1 Мur2Ас в составе продукта, имеет ту же конфигурацию, что и исходная гликозидная связь. Таким образом, данная реакция является реакцией замещения с сохранением конфигурации, а) Два механизма, предложенные для описания данной реакции. Слева изображен предложенный Филлипсом механизмSN1-типа. Изображенный справа механизм SN-типа лучше согласуется с современными экспериментальными данными, б) Поверхность активного центра лизоцима с шаростержневой моделью ковалентного фермент-субстратного промежуточного соединения. Боковые цепи в составе активного центра показаны как шаростержневые структуры, отходящие от ленточных структур (РDВ ID 1Н6М).
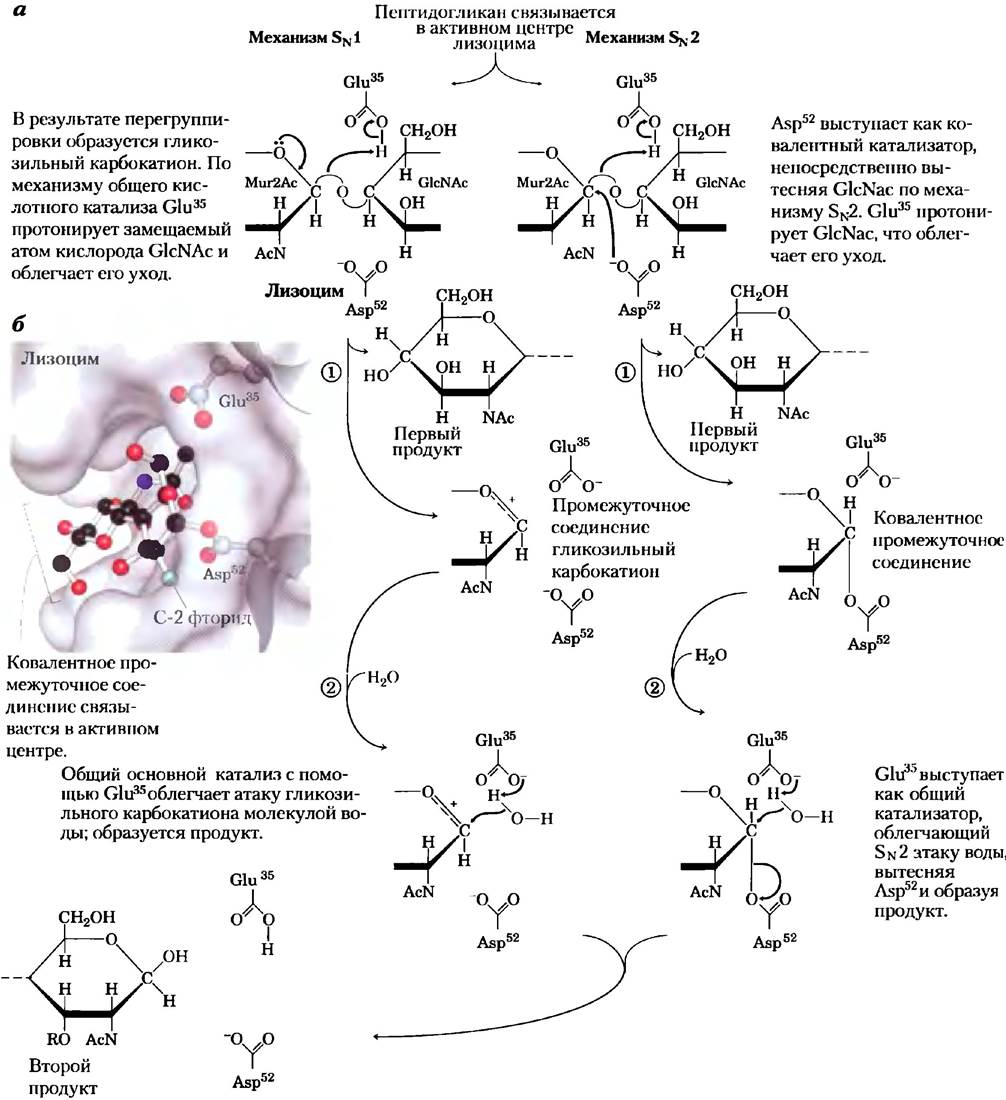
Казалось, что после идентификации аминокислотных остатков активного центра и детального изучения структуры белка в 1960-х гг. механизм действия лизоцима стал ясен. Однако окончательное понимание этого механизма пришло лишь почти через сорок лет. Существует два возможных пути протекания реакции, приводящих к образованию продуктов гидролиза гликозидной связи. Филлипс с сотрудниками предполагали, что реакция идет по диссоциативному SN 1 -типу (рис. 6-25, я, слева), при котором GlcNAcсначала диссоциирует (стадия 1) с образованием карбокатионного интермедиата. В данной схеме уходящий GlcNAc протонируется в соответствии с механизмом общего кислотного катализа при участии Glu35,расположенного в гидрофобном кармане, что сообщает его карбоксильной группе необычно высокое значение рКа. Карбокатион стабилизируется за счет резонансной структуры с участием кислорода шестичленного кольца, а также электростатических взаимодействий с отрицательным зарядом на соседнем Asp52. На стадии 2 вода атакует атом С-1 Мur2Ас, в результате образуется продукт. Альтернативный механизм (рис. 6-25, а, справа) включает в себя две последовательные стадии замещения SN2-типа. На стадии 1 Asp52 атакует атом С-1 Мгк2Ас и вытесняет GlcNAc. Как и в первом механизме, Glu35 выступает в качестве общей кислоты и протонирует уходящий GlcNAc. На стадии 2 вода атакует атом С-1 Mur2Ac, что приводит к вытеснению Asp52 и образованию продукта.
Механизм, предложенный Филлипсом (Sx1- типа), признавался большинством исследователей на протяжении более трех десятилетий. Однако сохранялись некоторые противоречия, и работа в данном направлении продолжалась. Методы анализа иногда развиваются слишком медленно, и бывает трудно поставить эксперимент, который бы прямо ответил на поставленный вопрос. Некоторые аргументы против гипотезы Филлипса ставили ее под сомнение, но не были особенно убедительны. Например, по оценкам, время полураспада гликозильного катиона должно было составлять 10-12 с, т. е. больше, чем период колебания молекулы, но недостаточно для диффузии других молекул. Еще более важно, что лизоцим является членом семейства ферментов, сохраняющих аномерную конфигурацию продукта такой же, какая была у субстрата (об аномерных конфигурациях углеводов см. гл. 7). Про все эти ферменты известно, что реакции с их участием протекают с образованием ковалентного промежуточного продукта, аналогичного тому, что мог бы возникать при альтернативном механизме действия лизоцима (SN2-типа). Таким образом, механизм Филлипса противоречил экспериментальным данным для близкородственных белков.
Эксперимент, проведенный Стефеном Уитерсом с сотрудниками в 2001 г., окончательно сдвинул чашу весов в пользу SN2-механизма. Используя мутантный фермент (в котором Glu33 был заменен Gin) и искусственный субстрат, что позволило снизить скорость ключевых стадий реакции, удалось стабилизировать ковалентное промежуточное соединение и изучить его напрямую с помощью методов масс-спектрометрии и рентгенокристаллографии (рис. 6-25, б).
Можно ли сказать, что механизм действия лизоцима доказан? Нет. Одной из особенностей научного подхода к решению проблемы, как говорил Альберт Эйнштейн, является то, что «никакое количество экспериментальных данных не может доказать, что данное утверждение верно, но всего лишь один эксперимент может доказать, что оно ошибочно». В случае механизма действия лизоцима кто-то может возразить (и уже возражали), что использование искусственных субстратов с атомами фтора у атомов С-1 и С-2, введенными для стабилизации ковалентного интермедиата, могло изменить путь реакции. Сильноэлектроотрицательный фтор мог дестабилизировать и без того электронодефицитный карбениевый ион интермедиата, возникший при реализации механизма SN1-типа. И тем не менее на сегодняшний день SN2-механизм лучше описывает существующие экспериментальные данные.
Понимание механизмов действия ферментов стимулирует развитие медицины
■ Большинство лекарств, применяемых для лечения различных болезней — от головной боли до ВИЧ-инфекции, представляют собой ингибиторы ферментов. Здесь мы рассмотрим два примера: антибиотик пенициллин (и его производные) и ингибиторы протеаз, используемые для лечения ВИЧ-инфекции; эти препараты являются необратимыми ингибиторами ферментов.
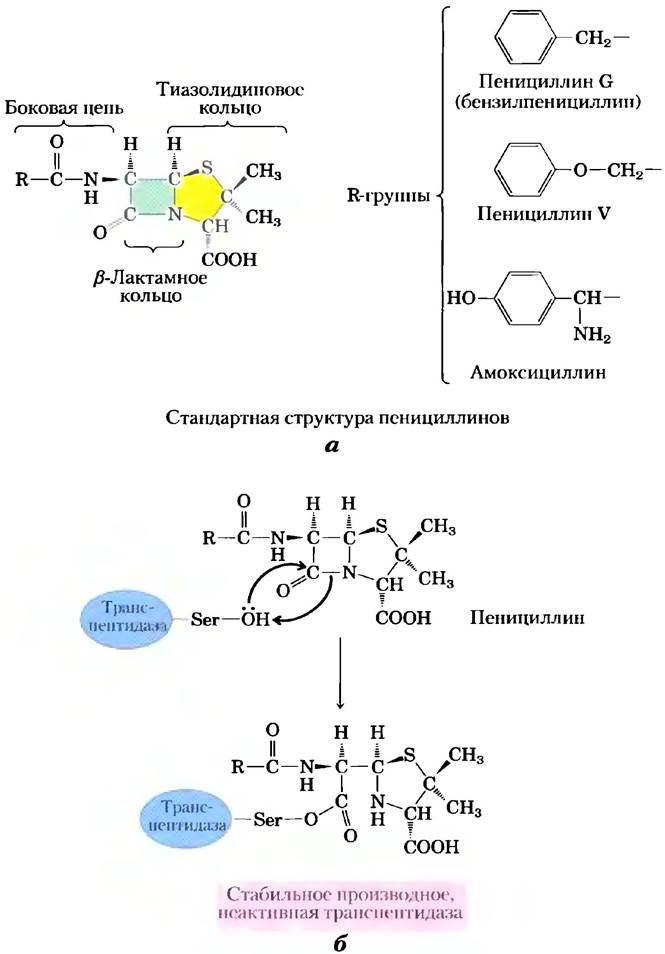
Пенициллин был открыт Александром Флемингом в 1928 г., но прошло еще 15 лет, прежде чем это сравнительно нестойкое вещество изучили настолько хорошо, что смогли использовать в качестве лекарства для лечения бактериальных инфекций. Пенициллин блокирует синтез пептидогликана (см. гл. 20; рис. 20-32) — основного компонента жесткой клеточной стенки, которая предохраняет бактерии от осмотического лизиса. Пептидогликан образуется в результате перекрестного сшивания полисахаридов и пептидов при участии фермента транспептидазы (рис. 6-26). Именно эту реакцию ингибируют пенициллин и его аналоги (рис. 6-27, а), имитирующие участок D-Ala- D-Ala в последовательности предшественника пептидогликана. Пептидная связь в предшественнике замещается реакционноспособным β-лактамным кольцом. При связывании пенициллина с транспептидазой серин активного центра фермента атакует карбонильную группу β-лактамного кольца, в результате чего образуется ковалентный аддукт пенициллина и фермента. Однако уходящая группа остается связанной с остатком β-лактамного кольца (рис. 6-27, б). Данный ковалентный комплекс необратимо инактивирует фермент. А это, в свою очередь, блокирует синтез клеточной стенки бактерии, и большинство бактерий погибают, поскольку их хрупкая внутренняя мембрана прорывается в результате осмотического шока.
Рис. 6-26. Реакция, катализируемая транспептидазой. Реакция связывания двух предшественников в более крупную полимерную молекулу пептидогликана происходит при участии остатка серина активного центра фермента в соответствии с механизмом ковалентного катализа, напоминающим механизм реакции, катализируемой химотрипсином. Интересно, что пептидогликан — одно из немногих природных соединений, в которых обнаружены остатки D-аминокислот. Серин активного центра атакует карбонильную группу пептидной связи между двумя остатками D-Аlа, образуя сложноэфирную связь между субстратом и ферментом и высвобождая концевой остаток D-Аlа. Далее аминогруппа второго предшественника атакует сложноэфирную связь, вытесняя фермент и сшивая тем самым два предшественника.

Рис. 6-27. Ингибирование транспептидазы β-лактамными антибиотиками, а) β-Лактамные антибиотики состоят из пятичленного тиазолидинового кольца, слитого с четырехчленным β-лактамным кольцом. Последнее является напряженной структурой и содержит амидный остаток, играющий важнейшую роль в остановке синтеза пептидогликана. В различных пенициллинах R-группа разная. Пенициллин G был выделен первым и до сих пор остается наиболее эффективным, однако он разрушается желудочным соком, и поэтому его вводят в организм путем инъекций. Пенициллин V почти так же активен и не разрушается в кислых условиях, поэтому его можно принимать перорально. Амоксициллин эффективен при широком спектре заболеваний, его можно принимать перорально, и поэтому его назначают наиболее часто из всех β-лактамных антибиотиков, б) Атака остатка Ser активного центра транспептидазы на амидную группу β-лактамного кольца приводит к образованию ацилированного фермента. Гидролиз этого соединения происходит настолько медленно, что реакция образования аддукта практически необратима, и транспептидаза остается в неактивной форме.
Использование человеком пенициллина и его производных привело к эволюции штаммов патогенных бактерий, которые начали синтезировать β-лактамазы (рис. 6-28, а) — ферменты, которые расщепляют и тем самым инактивируют β-лактамные антибиотики. В результате бактерии стали устойчивыми к действию антибиотиков этого класса. Гены этих ферментов быстро распространяются в популяции бактерий в условиях селективного давления, вызванного использованием (иногда чрезмерным) β-лактамных антибиотиков. На появление β-лактамаз фармацевты ответили созданием таких соединений, как клавулановая кислота — суицидный ингибитор, необратимо инактивирующий β-лактамазы (рис. 6-28, б). Структура клавулановой кислоты напоминает структуру β-лактамного антибиотика, и поэтому она образует ковалентный аддукт с остатком серина в активном центре β-лактамазы. Это приводит к определенным перестройкам и к образованию гораздо более реакционноспособного производного, которое затем подвергается атаке другим нуклеофилом активного центра, что приводит к необратимому ацилированию фермента и его инактивации. Широко применяемый лекарственный препарат Аугментин™ представляет собой комбинацию амоксициллина и клавулановой кислоты. «Химическая война» между человеком и бактериями продолжается. Были обнаружены штаммы болезнетворных бактерий, устойчивых как к амоксициллину, так и к клавулановой кислоте (это означает, что в молекуле β-лактамазы произошли мутации, предотвращающие ее взаимодействие с клавулановой кислотой). По всей видимости, производство новых антибиотиков в ближайшем будущем будет развиваться еще активнее.
Рис. 6-28. β-Лактамазы и их ингибиторы, а) β-Лактамазы участвуют в расщеплении β-лактамного кольца в β-лактамных антибиотиках, инактивируя их. б) Клавулановая кислота представляет собой суицидный ингибитор, действие которого основано на использовании нормального химического механизма образования в активном центре реакционноспособных частиц, с которыми взаимодействуют другие группы активного центра, что приводит к необратимому ацилированию фермента.

Антивирусные препараты — еще один пример современных лекарственных средств. Вирус иммунодефицита человека (ВИЧ) является причиной синдрома, приобретенного иммуннодефицита (СПИД). По оценкам, в 2005 г. от 37 до 45 млн человек были носителями вируса, причем ежегодно заражается от 3,9 до 6,6 млн человек, а умирает более 2,4 млн. Впервые о СПИДе как об эпидемии заговорили в 1980-х гг.
Вскоре был открыт вирус иммунодефицита, относящийся к группе ретровирусов. В геноме ретровирусов содержится РНК, а фермент обратная транскриптаза способен на основе РНК синтезировать комплементарную последовательность ДНК. Понимание механизмов действия ВИЧ и разработка методов лечения инфекции основываются на результатах многолетних научных исследований, проводившихся на других ретровирусах. Такой ретровирус, как ВИЧ, имеет сравнительно простой жизненный цикл (см. рис. 26-33). Его геномная РНК конвертируется в двойную спираль ДНК в результате действия обратной транскриптазы (см. гл. 26). Затем двухцепочечная ДНК встраивается в хромосому в ядре клетки хозяина при помощи фермента интегразы (см. гл. 25). Интегрированная копия вирусного генома может находиться в неактивном состоянии бесконечно долго. Но она также может транскрибироваться обратно в РНК, которая затем транслируется в белки, использующиеся для строительства новых вирусных частиц. Большая часть вирусных генов транслируется в длинные полипротеины, которые иод действием протеазы ВИЧ расщепляются на отдельные белки, необходимые для сборки вируса (см. рис. 26-34). В данном цикле принимают участие всего три важных фермента: обратная транскриптаза, интеграза и протеаза, и каждый из них является возможной мишенью для действия лекарственных препаратов.
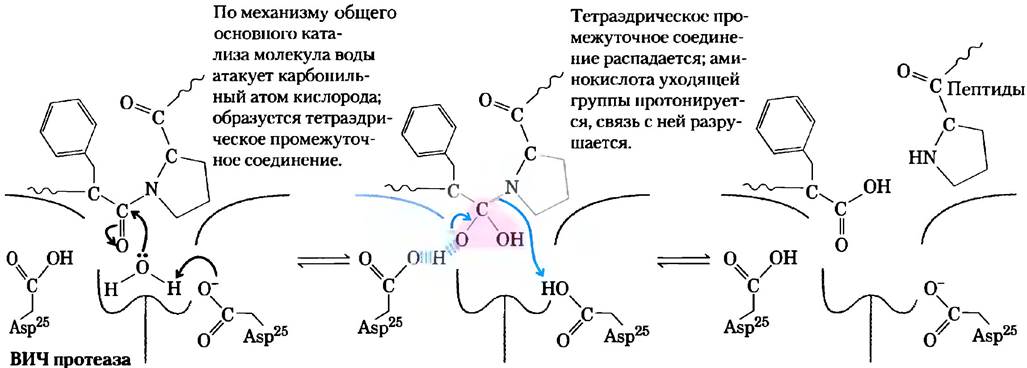
Существуют четыре основных подкласса протеаз. Сериновые протеиназы, такие как химотрипсин и трипсин, и цистеиновые протеиназы (в которых Cys играет приблизительно такую же роль в катализе, как и Ser) образуют ковалентные фермент-субстратные комплексы, а аспартильные протеиназы и металлопротеиназы таких комплексов не образуют. Протеаза ВИЧ является аспартильной протеиназой. Два остатка Asp в активном центре облегчают прямую атаку воды на расщепляемую пептидную связь (рис. 6-29). Сначала при атаке молекулы воды на карбонильную группу пептидной связи образуется неустойчивое тетраэдрическое производное, аналогичное тому, что мы видели в реакции химотрипсина. Это производное по структуре и энергетическому состоянию близко к переходному состоянию реакции. Лекарственные препараты, являющиеся ингибиторами протеазы ВИЧ, образуют нековалентные комплексы с ферментом, но связываются с ним настолько прочно, что их можно рассматривать в качестве необратимых ингибиторов. Прочность связывания частично объясняется тем, что эти вещества представляют собой аналоги переходного состояния (см. доп. 6-3). Мы указываем на этот факт, чтобы еще раз подчеркнуть, что принципы катализа, рассмотренные нами в настоящей главе, являются не просто абстрактными идеями. Их применение спасает жизнь людям.
Рис. 6-29. Механизм действия протеазы ВИЧ. Два остатка Asp активного центра (из разных субъединиц) действуют в качестве общих кислотно-основных катализаторов, облегчая атаку воды на пептидную связь. Неустойчивое тетраэдрическое промежуточное соединение, возникающее в ходе реакции, выделено розовым цветом.
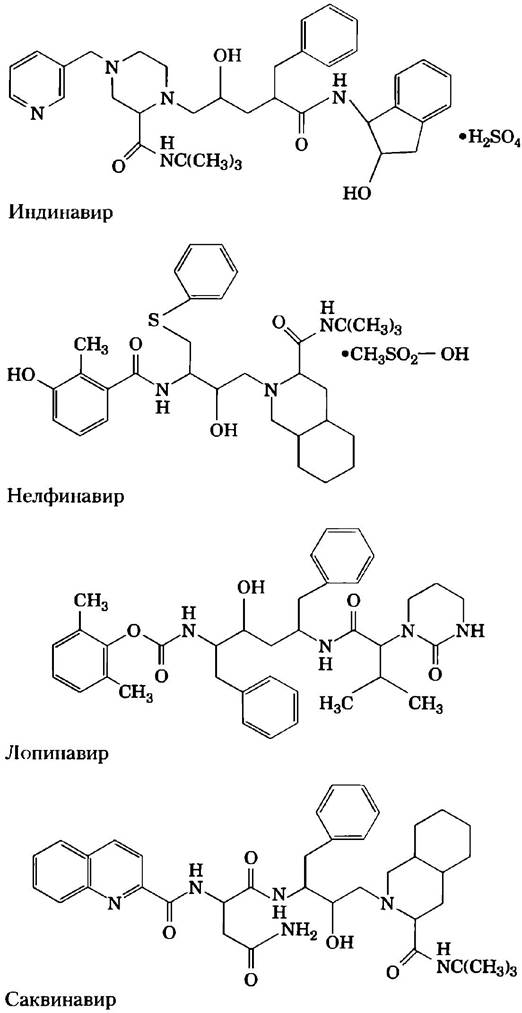
Протеаза ВИЧ наиболее эффективно расщепляет пептидные связи между остатками Phe и Pro. Активный центр фермента содержит карман, в котором может разместиться соседняя с расщепляемой связью ароматическая группа. Структура нескольких ингибиторов протеаз ВИЧ представлена на рис. 6-30. Эти структуры кажутся различными, но у них есть общая черта — гидроксильная группа в основной цепи, расположенная по соседству с боковой цепью, содержащей бензольную группу. За счет такого строения бензольное кольцо легко укладывается в карман для связывания ароматических групп. Соседняя гидроксильная группа имитирует отрицательно заряженный кислород в тетраэдрическом промежуточном продукте нормальной реакции, в результате чего образуется аналог переходного состояния. Остальная часть молекулы каждого ингибитора сконструирована таким образом, чтобы максимально соответствовать по форме тем углублениям, которые имеются на поверхности фермента, дополнительно усиливая связывание.
Рис. 6-30. Ингибиторы протеазы ВИЧ. Гидроксильная группа (выделена красным) имитирует атом кислорода в тетраэдрическом промежуточном продукте, создавая аналог переходного состояния. Соседнее бензольное кольцо (синее) помогает лекарству правильно расположиться в активном центре фермента.
Создание подобных препаратов значительно увеличило продолжительность жизни и улучшило качество жизни миллионов, пораженных спидом людей. ■
Краткое содержание раздела 6.4 Примеры ферментативных реакций
■ Химотрипсин — это сериновая протеаза, механизм действия которой хорошо изучен. Этот механизм включает в себя элементы общего кислотно-основного катализа, ковалентного катализа и стабилизацию переходного состояния.
■ Действие гексокиназы является прекрасным примером того, как при индуцированном соответствии используется энергия связывания субстрата.
■ Реакция, катализируемая енолазой, представляет собой пример катализа с участием ионов металлов.
■ Действие лизоцима основано на ковалентном и общем основном катализе и осуществляется в двух последовательных реакциях нуклеофильного замещения.
■ Понимание механизма действия ферментов позволяет создавать лекарства, которые являются ингибиторами этих ферментов.