ОСНОВЫ БИОХИМИИ ЛЕНИНДЖЕРА - ТОМ 1. ОСНОВЫ БИОХИМИИ СТРОЕНИЕ И КАТАЛИЗ - 2011
ЧАСТЬ I. СТРОЕНИЕ И КАТАЛИЗ
9. ТЕХНОЛОГИЯ НА ОСНОВЕ ИНФОРМАЦИИ ИЗ ДНК
9.2. От генов к геномам
Современная научная дисциплина геномика на сегодняшний день позволяет изучать ДНК в масштабе клетки, от отдельных генов до полного генетического комплекта организма — его генома. Геномные базы данных быстро растут по мере того, как завершается очередной этап секвенирования. Биология в XXI в. будет продвигаться с помощью информационных ресурсов, что всего несколько лет назад было даже трудно себе представить. Теперь обратимся к рассмотрению некоторых технологий, лежащих в основе такого прогресса.
Библиотеки ДНК представляют собой специализированные каталоги генетической информации
Библиотека ДНК представляет собой собрание ДНК-клонов, объединенных вместе в качестве источника ДНК для секвенирования и идентификации гена или изучения его функций. Библиотеки могут быть нескольких типов в зависимости от источника ДНК. Одной из самых больших библиотек ДНК является геномная библиотека; данные для нее получают при разрезании целого генома какого-то организма на тысячи фрагментов; все эти фрагменты клонируются путем их вставки в клонирующий вектор.
Первый шаг в получении геномной библиотеки — частичное расщепление ДНК эндонуклеазами рестрикции, так чтобы любая ее последовательность встречалась во фрагментах определенного диапазона длин — диапазона, совместимого с клонирующим вектором и гарантирующего присутствие практически всех последовательностей в клонах библиотеки. Слишком большие или слишком малые для клонирования фрагменты удаляются центрифугированием или электрофорезом. Клонирующий вектор, такой как плазмида ВАС или YAC, разрезается такой же эндонуклеазой рестрикции и лигируется вместе с геномными фрагментами ДНК. Совокупность связанных таким образом ДНК затем используется для трансформации бактериальных или дрожжевых клеток, образуя библиотеку типов клеток таким образом, что каждый тип несет в себе разную рекомбинантную молекулу ДНК. В идеальном случае вся ДНК в изучаемом геноме представлена в библиотеке. Каждая трансформированная бактериальная или дрожжевая клетка образует колонию, или «клон», идентичных клеток, причем каждая клетка включает в себя одну и ту же рекомбинантную плазмиду, одну из многих, представленных во всей библиотеке.
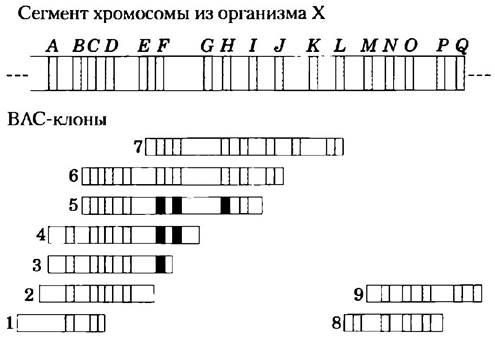
Используя методы гибридизации, исследователи могут упорядочить отдельные клоны в библиотеке, идентифицируя клоны с перекрывающимися последовательностями. Набор перекрывающихся клонов представляет собой каталог элементов протяженного сегмента генома и часто называется контигом (рис. 9-13). Установленные ранее нуклеотидные последовательности целых генов можно найти в библиотеке методами гибридизации, определяя, какие именно клоны в библиотеке содержат в себе искомую последовательность. Если же последовательность уже была картирована на хромосоме, то исследователи могут определить местонахождение (во всем геноме) клонированной ДНК и любого контига, частью которого она является. Хорошо организованные библиотеки могут содержать тысячи длинных контигов, относящихся к определенным хромосомам и упорядоченных в соответствии с ними, образуя подробную геномную карту. Известные последовательности в библиотеке (каждый из которых называется последовательно-меченным сайтом, или STS; от англ. sequence-tagged site) могут стать ориентирами для проектов по расшифровке геномов.
Рис. 9-13. Упорядочение клонов в библиотеке ДНК. Представлен сегмент хромосомы гипотетического организма X с маркерами от А до Q, изображающими последовательно-меченные сайты (STS — участки ДНК с известными нуклеотидными последовательностями, включая известные гены). Под хромосомой изображен набор упорядоченных ВАС-клонов с номерами от 1 до 9. Упорядочение клонов на генетической карте является многостадийным процессом. Присутствие или отсутствие STS в отдельном клоне можно определить гибридизацией — например, зондированием каждого клона ДНК, полученной из STS с помощью ПЦР. Как только все STS в каждом ВАС-клоне идентифицированы, клоны (и сами STS, если их местоположение еще не известно) можно расположить на карте в определенном порядке. Например, сравним клоны 3, 4 и 5. Маркер Е (синий) обнаружен во всех трех клонах; F (красный) в клонах 4 и 5, но не в 3; и G (зеленый) только в клоне 5. Это означает порядок следования сайтов Е, F, G. Клоны частично перекрываются и их очередность должна быть 3, 4, 5. Конечный упорядоченный набор клонов называется контигом.
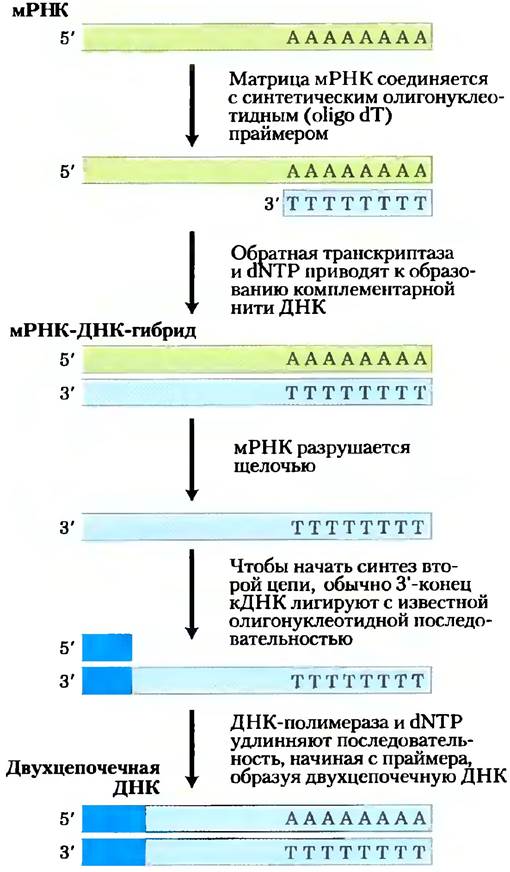
По мере того как все больше и больше нуклеотидных последовательностей генома становятся доступными, использование геномных библиотек постепенно убывает, и исследователи создают более специализированные библиотеки, предназначенные для изучения функций генов. В качестве примера можно привести библитотеку, которая включает только те гены, которые экспрессируются, т. е. транскрибируются в РНК, в данном организме или в определенных клетках или тканях. В таком банке нет некодирующих последовательностей, которые составляют значительную часть многих геномов эукариот. В первую очередь выделяют мРНК из организма или из его специфических клеток, а затем из РНК получает комплементарные ДНК (кДНК) в многостадийной реакции, которую катализирует фермент обратная транскриптаза (рис. 9-14). Получившиеся в результате фрагменты двухцепочечной ДНК затем помещают в подходящий вектор и клонируют, образуется популяция клонов под названием библиотека кДНК. Поиск определенного гена становится легче при обращении к библиотеке кДНК, образованной из мРНК клетки, когда известно, что данный ген экспрессируется. Например, если бы мы решили клонировать гены глобина, то вначале пришлось бы создать библиотеку кДНК из клеток- предшественников эритроцитов, в которых около половины мРНК кодируют глобины. Чтобы облегчить картирование больших геномов, кДНК в библиотеке можно частично секвенировать в произвольном порядке для образования полезного типа STS, названного меткой экспрессируемой последовательности (EST, от англ. expressed sequence tag). В диапазоне длин от нескольких десятков до сотен пар оснований можно расположить EST на карте большого генома, предоставляя, таким образом, маркеры экспрессируемых генов. Сотни тысяч EST нанесены на подробные геномные карты; они использовались в качестве ориентиров при расшифровке генома человека.
Рис. 9-14. Создание библиотеки кДНК из мРНК. мРНК клетки состоит из транскриптов тысяч генов, и образующиеся кДНК гетерогенны. Получаемая таким методом двойная цепь ДНК помещается в подходящий клонирующий вектор. Обратная транскриптаза может синтезировать ДНК на РНК- или ДНК-матрице (см. рис. 26-33).
Библиотеку кДНК можно сделать даже более специализированной, клонируя кДНК или фрагмент кДНК в вектор, в котором кДНК- последовательность соединяется с последовательностью маркера или сигнального гена; соединенные гены образуют «сигнальную конструкцию». Двумя подходящими маркерами являются гены зеленого флуоресцентного белка и эпитопные метки. Исходный ген, соединенный с геном зеленого флуоресцентного белка (GFP, от англ. greenfluorescent protein), приводит к образованию слитого белка, который чрезвычайно флуоресцентен — он буквально светится (рис. 9-15, а). Зеленый флуоресцентный белок (GFP) из медузы Aequorea victoriaимеет структуру β-бочонка, внутри которого находится флуорофор (см. доп. 12-3, с. 612). Флуорофор образуется в результате перегруппировки и окисления нескольких аминокислотных остатков в автокаталитической реакции, для которой требуется только молекулярный кислород (см. доп. 12-3, рис. 3). Поэтому этот белок легко клонировать в активной форме практически в любой клетке. Всего нескольких молекул белка достаточно, чтобы увидеть их под микроскопом; это позволяет следить за его перемещениями в клетке. Методом белковой инженерии удалось получить мутантные формы белка, способные светиться другими цветами и обладающие некоторыми другими свойствами (яркостью, стабильностью). Кроме того, несколько родственных белков были недавно выделены из других видов организмов.
Рис. 9-15. Специализированные библиотеки ДНК. а) Клонирование кДНК вблизи гена зеленого флуоресцентного белка (GFР) создает сигнальную конструкцию. В интересующем (вставка ДНК) и сигнальном генах происходит транскрипция РНК, а затем из мРНК-транскрипта получается слитый белок, GFР-часть которого видна в флуоресцентном микроскопе. На фотографии показан круглый червь, содержащий GFP-слитый белок, который проявляется только в четырех местах на его теле. Сигнальные конструкции б) Если кДНК клонируется вблизи гена эпитопной метки, получающийся слитый белок можно осадить антителами к эпитопу. Любые другие белки, которые взаимодействуют с меченым, также осаждаются, помогая объяснять белок-белковые взаимодействия.
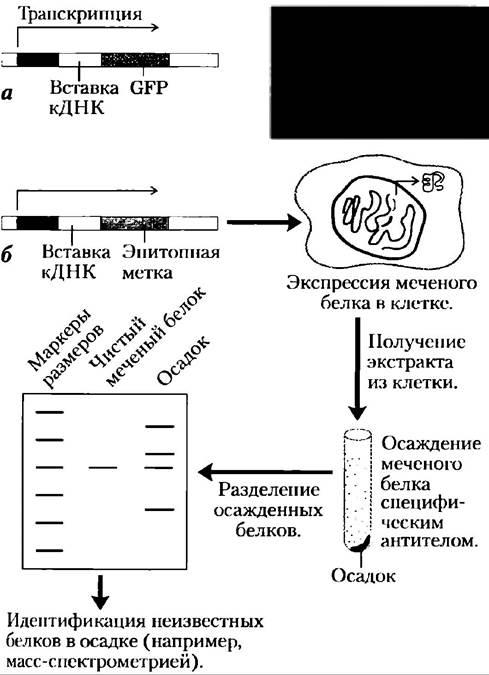
Эпитопная метка представляет собой короткую белковую последовательность, которая крепко связывается хорошо изученным моноклональным антителом (гл. 5). Меченый белок можно специфически осадить из общего белкового экстракта взаимодействием с антителом (рис. 9-15, б). Если еще какие-нибудь белки прикрепятся к меченому, то они также будут осаждены, предоставляя информацию о белок-белковых взаимодействиях в клетке. Разнообразие и практичность специализированных библиотек ДНК растут с каждым годом.
Полимеразная цепная реакция амплифицирует специфические последовательности ДНК
Проект «Геном человека» вместе с другими подобными попытками секвенирования геномов организмов любого вида обеспечивает беспрецедентный доступ к генетической информации. Это, в свою очередь, упрощает процесс клонирования отдельных генов для более детального биохимического анализа. Если известна нуклеотидная последовательность, но крайней мере, фланкирующих частей клонируемого сегмента ДНК, то можно значительно увеличить число копий такого сегмента, применив полимеразную цепную реакцию (ПЦР) — метод, созданный Кэри Маллис в 1983 г. Амплифицированную ДНК можно сразу клонировать или использовать во многих аналитических процедурах.
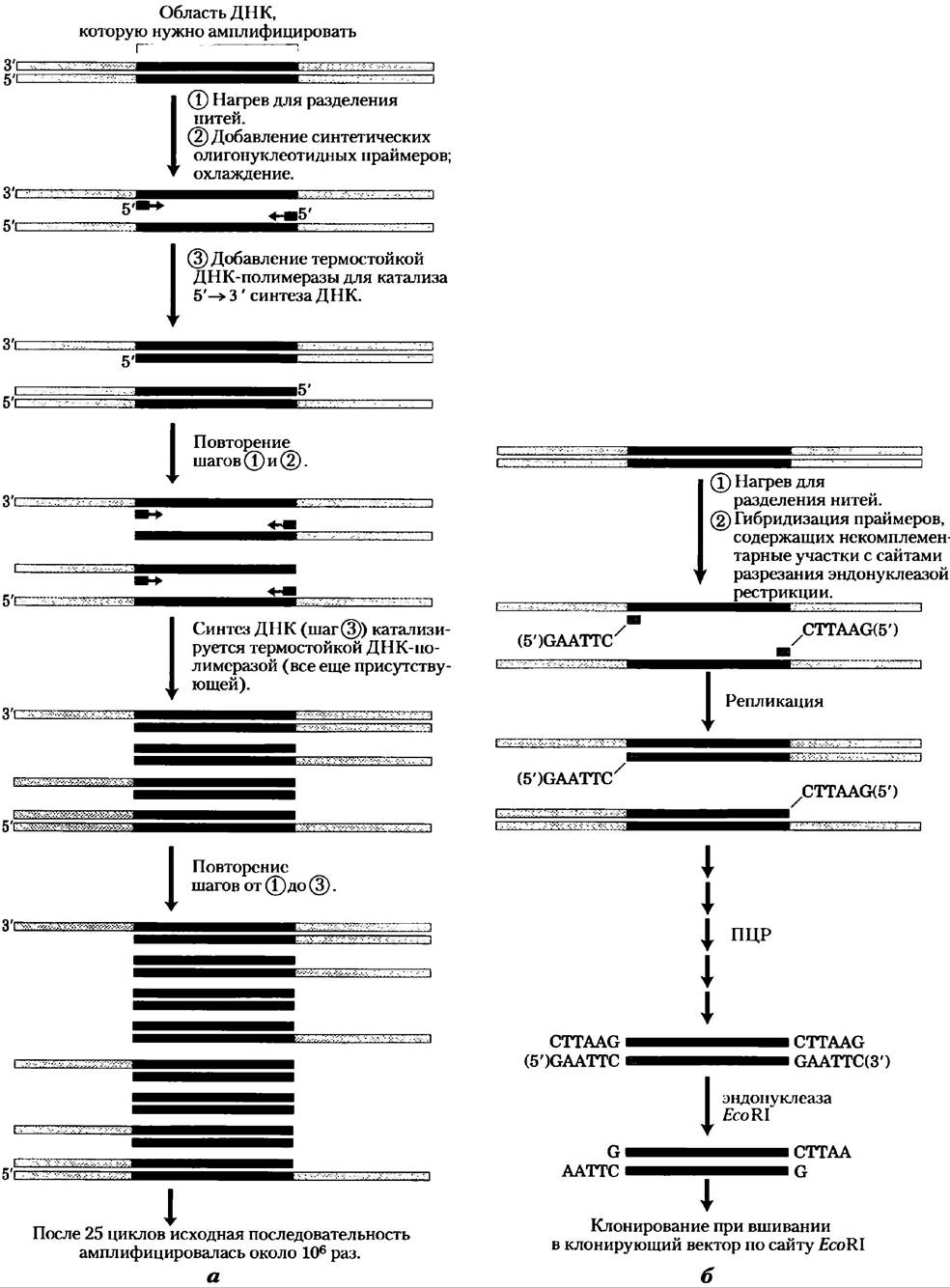
Метод ПЦР исключительно прост. Подготавливаются два синтетических олигонуклеотида, комплементарные последовательностям на противоположной цени ДНК прямо перед концами амплифицируемого сегмента. Олигонуклеотиды служат праймерами для репликации ДНК-полимеразой. 3'-Концы гибридизованных зондов направлены друг к другу и размещены для затравки синтеза ДНК по всему данному сегменту (рис. 9-16). (ДНК-полимеразы синтезируют нити ДНК из дезоксирибонуклеотидов, используя ДНК-матрицу; подробно об этом говорится в гл. 25) Выделенная ДНК, содержащая амплифицируемый сегмент, нагревается на короткое время для денатурации, а затем охлаждается в присутствии большого избытка синтетических олигонуклеотидных праймеров. Затем добавляются четыре типа дезоксинуклеозидтрифосфатов, и сегмент ДНК с праймером избирательно реплицируется. Цикл нагревания, охлаждения и репликации повторяется 25 или 30 раз в течение нескольких часов в ходе автоматизированного процесса, амплифицируя сегмент ДНК, ограниченный праймерами, до тех пор, пока его можно будет без труда анализировать или клонировать. В методе ПЦР используется термостабильная ДНК-полимераза, например, Taq-полимераза (полученная из бактерии, живущей при 90 °С), которая остается активной после каждого нагрева и не нуждается в замене. Тщательная разработка праймеров, используемых при ПЦР, например, включение участков разрезания эндонуклеазой рестрикции, может способствовать дальнейшему клонированию ам- плифицированной ДНК (рис. 9-16, б).
Рис. 9-16. Амплификация сегмента ДНК полимеразной цепной реакцией, а) Процедура ПЦР состоит из трех шагов. Нити ДНК (1) разделяются при нагревании, затем (2) гибридизуется в избытке коротких синтетических ДНК-праймеров (синие), которые ограничивают амплифицируемую область; (3) полимеризацией синтезируется новая ДНК. Эти три шага повторяют от 25 до 30 раз. Термостойкая ДНК-полимераза ТаqI (из Thermus aquaticus, бактериального вида, обитающего в термальных источниках) не денатурируется на этапах нагревания, б) ДНК, амплифицируемую с помощью ПЦР, можно клонировать. Праймеры могут содержать в себе некомплементарные концы, соответствующие сайтам разрезания эндонуклеазой рестрикции. Хотя эти части праймеров и не связываются с ДНК, в ходе ПЦР они включаются в амплифицируемую ДНК. Разрезание амплифицированных фрагментов в этих местах создает «липкие концы», используемые при вшивании амплифицированной ДНК в клонирующий вектор. Полимеразная цепная реакция
Эта технология очень чувствительна: с помощью метода ПЦР можно обнаружить и амплифицировать вплоть до одной молекулы ДНК практически в любом образце. Хотя ДНК и деградирует со временем (с. 415), ПЦР позволяет удачно клонировать ДНК из образцов возрастом более 40 000 лет. Исследователи применили эту технологию для клонирования фрагментов ДНК из мумифицированных останков людей и вымерших животных, например, мамонта; таким образом появились новые направления в молекулярной археологии и молекулярной палеонтологии. ДНК из мест обнаруженных захоронений амплифицировали с помощью ПЦР, а затем использовали, чтобы проследить миграции древнего человека. Эпидемиологи могут использовать образцы ДНК из человеческих останков, полученные методом ПЦР, для наблюдения за эволюцией патогенных вирусов человека. Помимо клонирования ДНК, метод ПЦР также эффективен в судебной медицине (доп. 9-1). Кроме того, ПЦР можно использовать для выявления вирусных инфекций прежде, чем они вызовут симптомы, а также для перенатальной диагностики генетических заболеваний.
Метод ПЦР также важен для выполнения задач секвенирования целого генома. Например, картирование экспрессируемых участков на определенной хромосоме часто включает амплификацию EST методом ПЦР, после чего следует гибридизация амплифицируемой ДНК с клонами упорядоченной библиотеки. Исследователи нашли также много других применений ПЦР для проекта «Геном человека», о котором сейчас и пойдет речь.
Мощное оружие при судебной экспертизе
Традиционно одним из самых точных способов для определения, находился ли человек на месте преступления, было снятие отпечатков пальцев. С разработкой технологии рекомбинантных ДНК появился еще более мощный метод — снятие отпечатка ДНК (этот метод также называют ДНК-типированием или ДНК- профилированием). Этот метод был впервые описан английским генетиком Алеком Джеффейсом в 1985 г.
Снятие отпечатка ДНК основывается на полиморфизмах нуклеотидной последовательности, т. е. незначительных отличиях последовательности (как правило, это одиночные изменения пар оснований) у разных людей, в среднем одной пары оснований на каждую 1000. Каждое отличие от прототипа геномной последовательности человека (впервые полученной) возникает у некоторой части человеческой популяции, и у каждого есть несколько таких отличий. Некоторые изменения в последовательности происходят на участках распознавания рестриктазами. Это приводит к различиям в размерах фрагментов ДНК, образующихся при разрезании соответствующей эндонуклеазой рестрикции. Такие отличия называются полиморфизмами длины фрагментов рестрикции(RFLP, от англ. restriction fragment length polymorphisms). Еще один тип последовательности, часто используемый для идентификации ДНК, — это короткие тандемные повторы(short tandem repeats, STR).
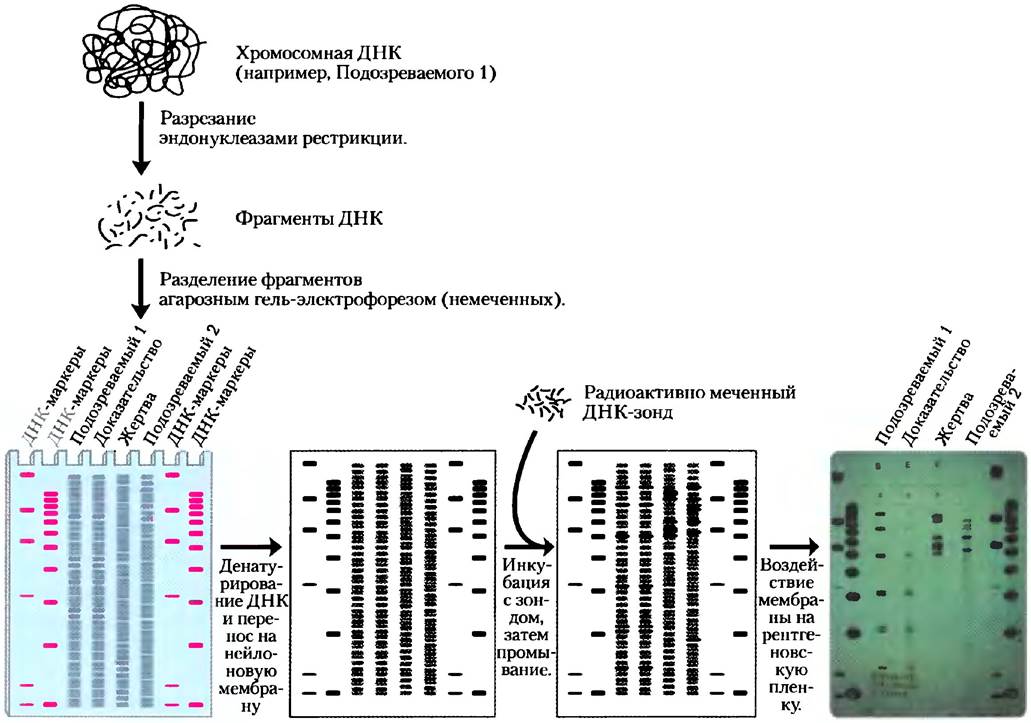
Обнаружение RFLP основывается на специальном методе гибридизации под названием саузерн- блоттинг (от англ. southern blotting; рис. 1). Фрагменты, полученные при разрезании геномной ДНК эндонуклеазами рестрикции, разделяют по размерам электрофорезом, денатурируют добавлением щелочи в агарозный гель, а затем переносят на нейлоновую мембрану для воспроизведения распределения фрагментов в геле. Мембрану погружают в раствор с радиоактивно меченным ДНК-зондом. Зонд для последовательности, которая неоднократно повторяется в геноме человека, обычно распознает несколько из тысяч фрагментов ДНК, образующихся при разрезании генома эндонуклеазой рестрикции. Авторадиография отображает фрагменты, с которыми гибридизуется зонд, так же, как и на рис. 1. Этот метод очень точный; он начал использоваться в судебной экспертизе в конце 1980-х гг. Однако для проведения анализа требуется много недеградированной ДНК (>25 нг). Такое количество ДНК часто невозможно обнаружить на месте преступления или катастрофы.
Рис. 1. Метод переноса по Саузерну для анализа полиморфизма длины рестрикционных фрагментов. Метод переноса по Саузерну, или саузерн-блоттинг, использующийся в молекулярной биологии для решения самых разных задач, назван по имени его создателя Джереми Саузерна. В данном примере из судебной практики ДНК из спермы, обнаруженной на теле изнасилованной и убитой жертвы, сравнивали с образцами ДНК жертвы и двух подозреваемых. Каждый образец ДНК расщепили на фрагменты и разделили в геле методом электрофореза. Для идентификации фрагментов использовали радиоактивно-меченный ДНК-зонд, комплементарный участку последовательности определенного фрагмента. Размеры идентифицированных фрагментов ДНК жертвы и двух подозреваемых различались между собой. Как видно, расположение полос ДНК одного из подозреваемых идентично таковому для образца ДНК, взятой на месте преступления.
В более чувствительных методах идентификации ДНК используются возможности полимеразной цепной реакции (ПЦР; см. рис. 9-16), а также анализ STR. Короткий тандемный повтор — это короткая последовательность ДНК, повторяющаяся несколько раз в определенных участках хромосомы; чаще всего последовательность содержит четыре пары нуклеотидов. Наиболее полезные для идентификации участки ДНК обычно содержат от 4 до 50 повторов (всего от 16 до 200 пар нуклеотидов в случае тетрануклеотидных повторов) и в человеческой популяции могут иметь разную длину. В геноме человека охарактеризовано свыше 20 000 тетрануклеотидных повторов. По оценкам, всего в геноме человека может содержаться свыше миллиона STR различных типов, что составляет около 3% всей геномной ДНК.
Таблица 1. Характеристика участков, использованных для базы CОDIS
Локус |
Хромосома |
Повтор |
Длина повтора (разброс)* |
Число изученных аллелей** |
CSF1PO |
5 |
TAGA |
5-16 |
20 |
FGA |
4 |
СТТТ |
12,2-51,2 |
80 |
ТН01 |
11 |
ТСАТ |
3-14 |
20 |
ТРОХ |
2 |
GAAT |
4-16 |
15 |
VWA |
12 |
[TCTG][TCTA] |
10-25 |
28 |
D3S1358 |
3 |
[TCTG][TCTA] |
8-21 |
24 |
D5S818 |
5 |
AGAT |
7-18 |
15 |
D7S820 |
7 |
GATA |
5-16 |
30 |
D8S1179 |
8 |
[TCTA][TCTG] |
7-20 |
17 |
D13S317 |
13 |
TATC |
5-16 |
17 |
D16S539 |
16 |
GATA |
5-16 |
19 |
D18S51 |
18 |
AGAA |
7-39,2 |
51 |
D21S11 |
21 |
[TCTA][TCTG] |
12-41,2 |
82 |
Amelogenin |
X,Y |
Неприменимо |
* Длина повторов в человеческой популяции. В некоторых аллелях могут встречаться частичные или неточные повторы.
** Число разных аллелей в человеческой популяции, изученных на данный момент. Тщательный анализ локусов многих людей — необходимое условие для использования ДНК-типирования в криминалистике.
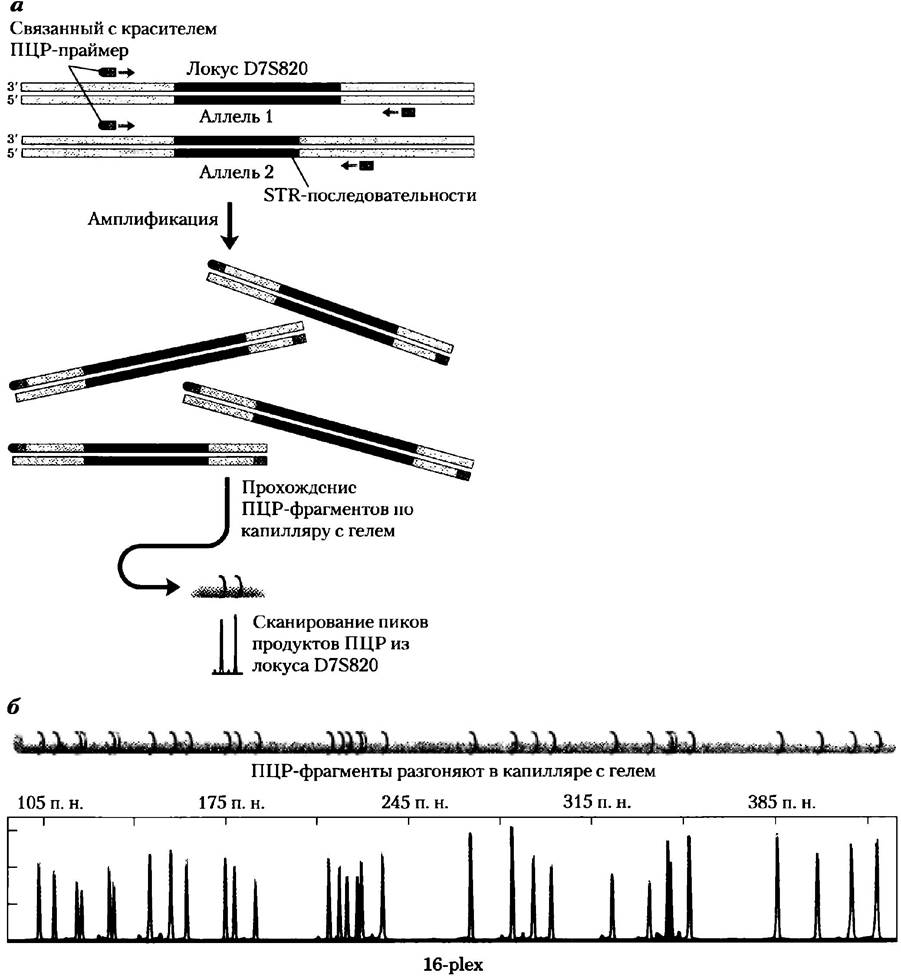
В анализе STR применяют полимеразную ценную реакцию. В судебной практике уже давно (в начале 1990-х гг.) стало очевидным преимущество анализа STR по сравнению с анализом полиморфизма длины рестрикционных фрагментов в связи с более высокой чувствительностью метода. Последовательности ДНК, ограничивающие STR, уникальны для каждого типа повтора и идентичны (за исключением очень редких мутаций) у всех людей. Для ПЦР используют праймеры, связывающиеся с этими последовательностями и позволяющими амплифицировать ДНК тандемного повтора (рис. 2, а). Таким образом, длина продукта ПЦР соответствует длине тандемного повтора в образце. Поскольку каждый человек наследует по одной хромосоме в паре от каждого родителя, длины тандемных повторов на двух хромосомах часто различны, в результате чего при анализе ДНК одного человека получают два сигнала. Если анализировать участки STR, расположение полос, получаемое при снятии отпечатков ДНК, индивидуально для каждого человека. Метод ПЦР позволяет исследователям получать отпечатки из ДНК массой менее 1 нг, даже частично разрушенной, например, из единственного волосяного фолликула, капли крови, небольшого количества спермы, найденной на жертве изнасилования, или из образцов ДНК давностью в несколько месяцев или даже лет.
Рис. 2. ПЦР-анализ локусов STR. а) Праймеры для ПЦР сконструированы таким образом, чтобы амплифицировать фрагмент ДНК, содержащий повтор. Один из двух праймеров связан с флуоресцентным красителем (зеленый кружок). Две хромосомы человека могут иметь различные аллели, с разным числом повторов в одном и том же локусе. Поэтому в результате проведения ПЦР могут образоваться два продукта слегка различающегося размера. Эти продукты разделяют с помощью очень тонкого полиакриламидного геля в капиллярной трубке. Образующиеся флуоресцентные полосы анализируют с помощью прибора и получают набор пиков. При сравнении этого набора с маркерами удается определить размер каждого продукта ПЦР и, следовательно, длину STR соответствующего аллеля. б) Сочетание продуктов ПЦР от нескольких локусов создает сложный рисунок, как тот, что изображен здесь. (В данном случае это коммерческий набор для анализа STR, содержащий последовательности из 16 локусов, так называемый 16-рlех.) Для такого анализа нужно несколько наборов праймеров — по одному набору для каждого локуса. Для облегчения идентификации локусов праймеры связаны с метками различного цвета. Кроме того, ПЦР-праймеры для каждого локуса подобраны таким образом, чтобы диапазон размеров получающихся продуктов отличался как можно сильнее от тех, что получаются с другими праймерами.
Для успешного использования анализа коротких тандемных повторов в судебной практике необходимы стандарты. Первый такой стандарт был разработан в Великобритании в 1995 г. Американский стандарт, названный COmbined DNA Index 5ystem (CODIS), был введен в 1998 г. Система CODIS использует 13 хорошо изученных участков STR (табл. 1); этот контроль обязательно должен быть включен в любой эксперимент по идентификации ДНК, проводимый в США. В качестве маркера также используется ген амелогенина. Фланкирующие последовательности этого гена, расположенного на половых хромосомах человека, на X- и Y-хромосоме несколько различаются. Поэтому при амплификации гена амелогенина методом ПЦР образуются продукты разного размера, что позволяет определить пол донора ДНК. База данных CODIS к 2006 г. содержала 2,8 млн образцов и была доступна повсеместно (во всех штатах). По данным на 2005 г., к этой базе данных обращались при более чем 25 000 судебных разбирательств.
Существуют удобные наборы, позволяющие амплифицировать 16 и более локусов STR в одной пробирке. В состав таких наборов (рис. 2, б) входят уникальные для каждого локуса ПЦР-праймеры. Каждый праймер синтезирован таким образом, чтобы не происходило гибридизации с другим праймером в данном наборе, и чтобы в ПЦР образовывались продукты разного размера, что позволяет в процессе электрофореза разнести сигналы от разных локусов. Праймеры связаны с красителями, помогающими различить продукты ПЦР. Наиболее широко применяющийся в настоящее время набор содержит 13 локусов CODIS, амелогенин и два дополнительных локуса (т. е. всего 16), используемые во всем мире при проведении судебных экспертиз. Такие наборы чрезвычайно полезны для установления личности человека. При наличии хорошего профиля ДНК вероятность совпадения сигналов для двух людей во всей человеческой популяции меньше 1:108.
ДНК-типирование используется как при осуждении, так и при оправдании подозреваемых, а также в иных случаях для установления источника с очень высокой степенью достоверности. Влияние таких процедур на судопроизводство будет продолжать расти, поскольку организации договариваются о стандартах, а методы становятся повсеместно официально принятыми в криминалистических лабораториях. Даже загадки убийств, совершенных несколько десятилетий назад, можно раскрыть: в 1996 г. получение отпечатков ДНК помогло подтвердить идентификацию костей последнего русского царя и его семьи, убитых в 1918 г.
На основе нуклеотидных последовательностей генома создаются самые большие генетические библиотеки
Геном является самым большим источником информации об организме, и нет такого генома, который интересовал бы нас больше нашего собственного. Менее чем через 10 лет после разработки практических методов секвенирования ДНК начались серьезные обсуждения перспектив секвенирования всех 3 млрд пар нуклеотидов генома человека. Международный проект «Геном человека» стартовал после выделения существенного финансирования в конце 1980-х гг. В конечном итоге, в развитие проекта значительные вклады внесли 20 секвенирующих центров в шести странах: США, Великобритании, Японии, Франции, Китае и Германии. Основная координация осуществлялась Управлением исследования генома в Национальном институте здоровья (США) вначале под руководством Джеймса Уотсона, а после 1992 г. — Френсиса Коллинза. Вначале задача определения последовательности 3 • 109 п. н. генома казалась титанической работой, но постепенно она стала приводить к технологическим успехам. Полная последовательность генома человека была опубликована в апреле 2003 г., на несколько лет раньше намеченного срока.
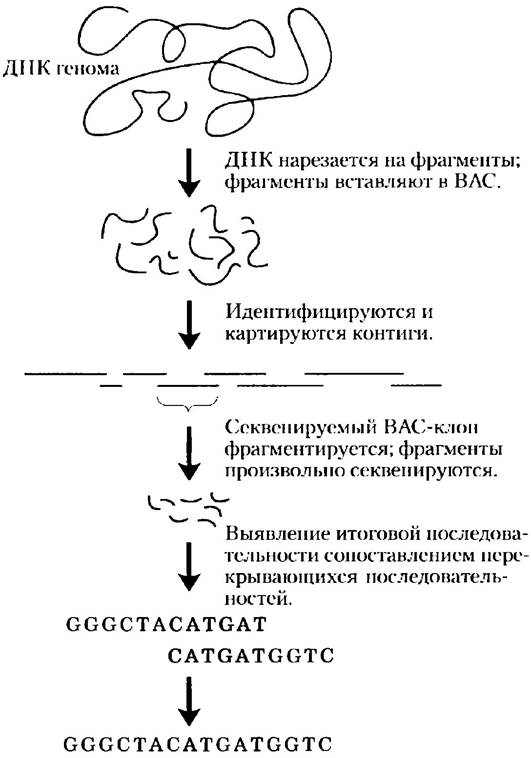
Это достижение стало результатом тщательно спланированной международной работы на протяжении 14 лет. Исследовательские группы впервые создали подробную карту генома человека, причем клоны, полученные из каждой хромосомы, организовывались в серию длинных контигов (рис. 9-17). Каждый контиг содержал ориентиры в форме STSна расстоянии менее 100 000 и. н. Картированный таким образом геном можно было поделить среди международных секвенирующих центров, причем каждый центр секвеи провал картированные BAC- или YAC-клопы, соответствующие своим определенным сегментам генома. Из-за того, что многие клоны имели длину более 100 000 и. н., а методы секвенирования могли распознать всего лишь от 600 до 750 п. н. нуклеотидной последовательности за раз, каждый клон надо было секвенировать по частям. Стратегия секвенирования основывалась на принципе shotgun («выстрела из дробовика»), в котором исследователи использовали новые мощные секвенаторы для определения последовательностей произвольных сегментов данного клона, а затем объединяли их с помощью компьютерной идентификации перекрываний. Число секвенированных образцов клона подсчитывалось статистически, так что весь клон целиком определялся в среднем 4-6 раз. Затем из баз данных брали секвенированную ДНК для сборки целого генома. Создание генетической карты было трудоемкой задачей, его ход сопровождался ежегодными отчетами в основных журналах на протяжении 1990-х гг., к концу десятилетия карта была составлена почти полностью. Завершение всего проекта по определению нуклеотидной последовательности генома человека первоначально было запланировано на 2005 г., но материальные и технические вложения ускорили процесс.
Рис. 9-17. Стратегия проекта «Геном человека». Полученные из геномной библиотеки клоны размещались в соответствующем порядке на подробной генетической карте, а затем отдельные клоны секвенировались методом shotgun. Методики секвенирования, применяемые для коммерческих целей, позволили отказаться от создания генетической карты и секвенировать целый геном клонированием по методу shotgun.
Коммерческий вклад в расшифровку генома человека был инициирован только что организованной в 1997 г. Celera Corporation. Под руководством Дж. Крэйга Вентера эта группа воспользовалась иной стратегией под названием «секвенирование целого генома методом shotgun», который исключил этап сборки карты генома. Вместо этого были секвенированы сегменты ДНК, взятые произвольным образом из всего генома. Секвенированные сегменты упорядочивались путем компьютерной идентификации перекрывающихся последовательностей (при этом обращений к подробной геномной карте государственного проекта было немного). В начале проекта «Геном человека» секвенирование методом shotgun в таких масштабах казалось далеким от практической реализации. Однако усовершенствования компьютерного программного обеспечения и автоматизации секвенирования сделали к 1997 г. такой подход осуществимым. Соревнование между частным и государственным вкладами в секвенирование существенно сэкономило время до окончания проекта. За публикацией в 2006 г. предварительных данных о нуклеотидной последовательности генома человека последовали два года завершающих работ по устранению примерно тысячи нестыковок, чтобы предоставить высококачественные данные о последовательности, которые согласовались бы между собой по всему геному.

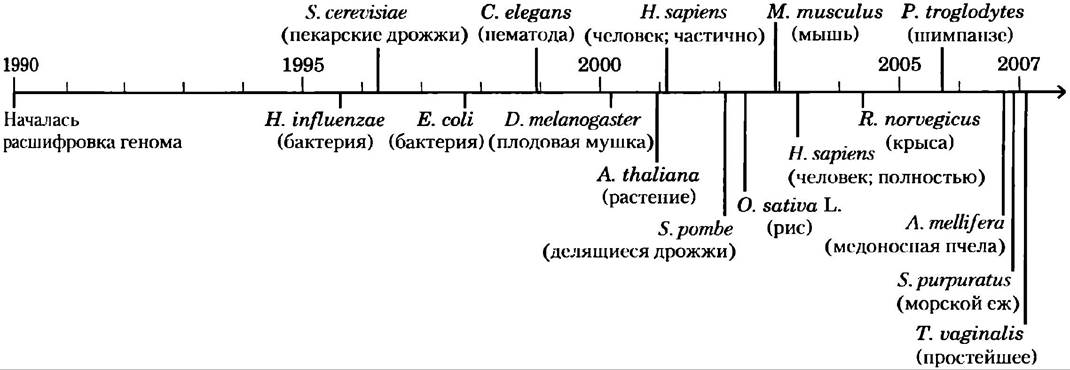
Проект «Геном человека» ознаменовал кульминацию биологии XX в. и предвещает совсем иную науку в следующем веке. Геном человека является всего лишь одним из этапов, поскольку наряду с ним расшифровываются (или уже расшифрованы) геномы многих других видов, в том числе дрожжей Saccharomyces cerevisiae (расшифровка завершена в 1996 г.) и Schizosaccharomyces pombe (2002), нематоды Cae.norhabditis elegans (1998), плодовой мушки Drosophila melanogaster (2000), растения Arabidopsis thaliana (2000), мыши Mus musculus (2002), рыбы-зебры и множества видов бактерий и архей (рис. 9-18). Ранние попытки расшифровки генома были сфокусированы в основном на видах, обычно используемых в лабораторных целях. Технология развивается, так что к моменту выхода этой книги станут известны полные последовательности геномов свыше 1200 организмов самых разных видов. Сегодня уже предпринимаются широкомасштабные попытки картирования генов, обнаружения новых белков и генов, вызывающих заболевания, а также многие другие проекты.
Рис. 9-18. Временная шкала секвенирования генома. Дискуссии в середине 1980-х гг. привели к тому, что начало проекта состоялось только в 1989 г. Предварительная работа, включавшая составление полной генетической карты и предоставление геномных ориентиров, заняла большую часть 1990-х гг. Были запущены также отдельные проекты по секвенированию геномов других организмов, необходимых для иследовательской практики. В настоящее время завершена расшифровка геномов многих видов бактерий (например, Haemophilus influenzae), дрожжей (S. cerevisiae), нематоды (С. elegans), насекомых (D. melanogaster и Apis mellifera) и растений {A. thaliana и Oryza sativa L), грызунов (Mus musculus и Rattus norvegicus), приматов (Homo sapiens и Pan troglodytes) и некоторых возбудителей половых инфекций человека (например. Trichomonas vaginalis). У каждого геномного проекта есть сайт в Интернете, который служит центральным хранилищем данных.
В результате исследований была создана база данных с таким потенциалом, который может не только привести к быстрому прогрессу биологии, но и изменить само представление людей о самих себе. Первые наши впечатления от расшифровки генома человека колебались от ощущения запутанности до осознания глубокой мудрости природы. Мы не настолько сложны, как думаем об этом. Оценки, сделанные несколько десятков лет назад, что у человека примерно 100 000 генов, распределенных среди 3,2 • 109 и. н. генома, не оправдались: у нас всего лишь от 25 000 до 30 000 генов. Это, судя по всему, в полтора раза больше, чем у дрозофилы (20 000 генов), и несколько больше, чем у нематоды (23 000). Несмотря на то что человек появился в ходе эволюции сравнительно недавно, геном наш очень стар. Из 1278 семейств белков, обнаруженных в одном древнем слое, только 94 были уникальными для позвоночных. Однако хотя многие типы белковых доменов у нас общие с растениями, червями и плодовыми мушками, но используем мы их в более сложных механизмах. Альтернативные пути экспрессии генов (гл. 26) дают возможность получения более одного белка с одного гена; этот процесс встречается у людей и других позвоночных чаще, чем у бактерий, червей или любых других форм жизни. Это способствует большей сложности белков, образующихся из нашего генного комплекта.
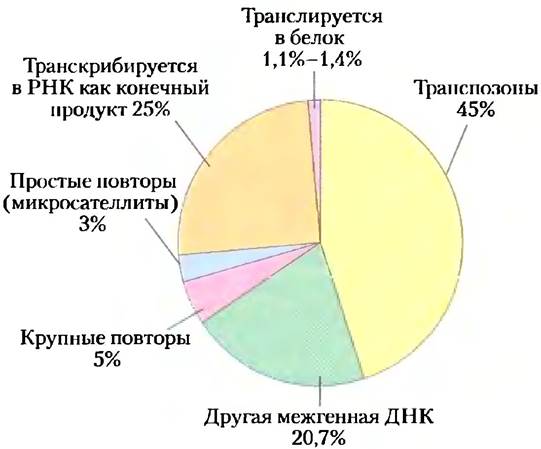
Теперь стало известно, что только от 1,1% до 1,4% нашей ДНК действительно кодирует белки (рис. 9-19). Больше 50% нашего генома состоит из коротких повторяющихся последовательностей, подавляющее большинство которых — около 45% нашего генома в целом — происходят из транспозонов, небольших мобильных последовательностей ДНК, являющихся молекулярными паразитами (гл. 25). Большинство транспозонов находились в нашем геноме продолжительное время и теперь настолько изменились, что больше не могут перемещаться на новые участки. Остальные все еще активно двигаются с низкой периодичностью, делая геном динамичным и развивающимся. Наконец, немногочисленные транспозоны были приняты своим хозяином и, по-видимому, выполняют важные клеточные функции.
Рис. 9-19. Структура генома человека. На диаграмме показаны доли различных видов последовательностей в нашем геноме.
Что же можно сказать на основе всей этой информации о том, насколько один человек отличается от другого? В пределах человеческой популяции существуют миллионы отличий на одно основание ДНК, называемых однонуклеотидными полиморфизмами, или SNPs (от англ. single nucleotide polymorphisms, произносится как «снипсы»). Каждый человек отличается от другого одной парой оснований на каждую тысячу. Из этих небольших генетических отличий возникает разнообразие людей, о котором мы все знаем, — различия в цвете волос и глаз, аллергии к лекарствам, размере стопы и даже (до некоторой неизвестной степени) в поведении. Некоторые SNPs связаны с определенными человеческими популяциями и могут дать важную информацию о миграциях человека, которые происходили тысячи лет назад, и о нашем более отдаленном эволюционном прошлом.
Как эта информация помогает нам понять, что именно делает человека человеком? Ответы па некоторые вопросы можно найти на основании анализа генома нашего ближайшего родственника шимпанзе. Геномы человека и шимпанзе различаются по нуклеотидному составу лишь на 1,2%, а по составу генов, кодирующих белки, различие еще меньше. Кажется, что это немного, однако в таких больших геномах это различие составляет около 35 млн п. н., еще 5 млн коротких вставок или делеций и довольно большое число более протяженных геномных перестановок. Далеко не простое дело — выяснить, какие же из этих различий ответственны за отличия человека от шимпанзе на фенотипическом уровне. Анализ генома приматов может оказать значительную помощь в понимании биохимической организации и эволюции человека, однако решение этой задачи пока находится в самом начале пути.
Насколько бы впечатляющим ни был успех, расшифровка генома человека сама по себе не так трудна по сравнению с тем, что следует сделать дальше — попытаться осознать всю информацию, хранящуюся в геноме. Геномные последовательности, ежемесячно добавляемые в международную базу данных, являются «дорожными картами», части которых написаны на языке, который мы еще не знаем. Но они приносят огромную пользу, способствуя открытию новых белков и процессов, оказывающих влияние на каждый аспект биохимии, и это станет ясно из следующих глав.
Краткое содержание раздела 9.2 От генов кгеномам
■ Наука геномика занимается активным изучением геномов и их содержимым.
■ Сегменты ДНК генома можно собрать в библиотеки — например, генные банки и библиотеки кДНК — с огромным диапазоном целей и задач.
■ Для амплификации отдельных сегментов ДНК из библиотеки ДНК или целого генома можно использовать полимеразную цепную реакцию (ПЦР).
■ Объединенным усилием международных исследовательских коллективов удалось полностью расшифровать геномы многих организмов, включая человека, и информация о них теперь доступна в публичных базах данных.