Основы биоинформатики - Огурцов А.Н. 2013
Информационные принципы в биотехнологии
Секвенирование биологических последовательностей и экспрессия генов
Экспрессия генов
Экспрессия генов - это процесс, в котором наследственная информация от гена (последовательности нуклеотидов ДНК) преобразуется в функциональный продукт - РНК или белок. Фактически ген при экспрессии используется как своего рода план синтеза определённого белка.
Картины экспрессии гена дают ключи к раскрытию его биологической роли. Все функции клеток, тканей и органов управляются дифференциальной экспрессией генов.
Анализ экспрессии гена проводят с целью изучения его функции. Информация о том, какие гены экспрессируются в здоровых и больных тканях, позволяет определить как набор белков, характерный для нормальной функции, так и отклонения состава белков, соответствующие заболеваниям.
Эти данные затем используются в разработке новых диагностических тестов различных заболеваний, а также новых лекарств, способных влиять на активность поражённых генов или белков.
Прежде экспрессию генов изучали на уровне РНК или белка, по принципу ген-за-геном, с помощью анализа методами Нозерн и Вестерн блоттинга (см. [7], п. 11.4). Теперь известны способы анализа общей экспрессии, в которых все гены исследуют одновременно.
Простой, но относительно дорогой методикой анализа на уровне РНК является прямая выборка последовательностей из наборов РНК, или библиотек кДНК, или даже из баз данных последовательностей.
В более совершенной методике, получившей название SAGE (Serial Analysis of Gene Expression - серийный анализ экспрессии генов), от каждой кДНК синтезируют очень короткие ярлыки последовательности (обычно 8-15 нуклеотидов), после чего их соединяют вместе по несколько сотен и таким образом формируют сцепку ярлыков для начала секвенирования. В одной реакции секвенирования может быть получена информация об относительном содержании сотен различных мРНК. Каждый ярлык SAGE уникально обозначает каждый ген, и путём подсчёта числа ярлыков могут быть определены относительные уровни экспрессии каждого гена.
Наиболее производительным является анализ экспрессии генов с помощью микроматриц ДНК.
Микроматрица ДНК - это матрица нитей ДНК (часто называемых признаками или ячейками), размещённых на миниатюрной подложке из нейлонового фильтра или предметного стекла (см. [7], п. 13.5).
Каждый элемент такой матрицы представляет собой множество тождественных одинарных нитей ДНК, представляющих определённый ген.
Явление гибридизации ДНК позволяет с помощью ДНК-зондов выделить молекулы ДНК из очень сложной смеси, например, из набора элементов целой молекулы ДНК или клеточной РНК. ДНК-зонд - это отдельная молекула ДНК (или РНК в случае РНК-зонда), с которой ковалентно связана радиоактивная или флуоресцентная метка.
Матрицу обычно гибридизируют комплексным зондом РНК; такой зонд производят путём мечения совокупной смеси молекул РНК, полученных из клетки определённого типа. Таким образом, состав зонда отражает относительное число отдельных молекул РНК в клетке-источнике.
Если выполняется ненасыщаемая гибридизация, то интенсивность сигнала каждого элемента микроматрицы представляет относительное содержание соответствующей РНК в зонде и, следовательно, позволяет одновременно визуализировать относительные уровни экспрессии нескольких тысяч генов.
Наиболее широко применяют метод с автоматизированным нанесением отдельных клонов ДНК на подложку (покрытое специальным составом предметное стекло), например, с помощью струйного принтера (ink-jet p rinting, IJP). Такие IJP-матрицы ДНК могут иметь плотность до 5000 элементов на квадратный сантиметр. Элементы содержат денатурированные перед началом гибридизации молекулы ДНК (клоны из исследуемого генома или молекулы кДНК) до 400 bp (рисунок 71).
Сначала клоны ДНК размножают и наносят на подложку, в результате чего получают микроматрицу (рисунок 71(a)). Одновременно эталонные фрагменты РНК и исследуемые фрагменты РНК образца обратно транскрибируют в ДНК-фрагменты и метят различными флуоресцентными красителями, которые высвечивают в разных областях спектра (например, красной и зелёной). Затем эти флуоресцентные зонды гибридизируют с ДНК на микроматрице (рисунок 71(a)). После этого микроматрицу промывают водой, чтобы удалить негибридизировавшиеся зонды, при помощи лазерного возбуждения измеряют интенсивность флуоресценции каждого красителя на всех элементах (генах) микроматрицы и преобразуют эти данные в относительные уровни экспрессии генов в исследуемом образце по сравнению с эталоном.
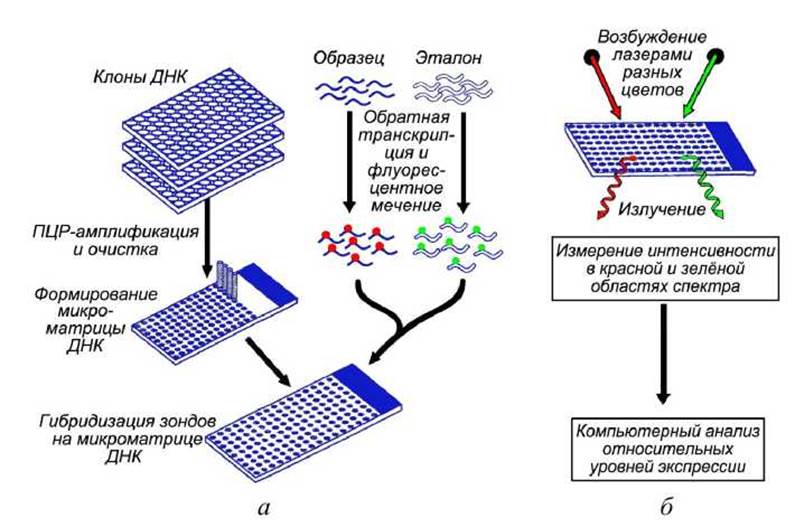
Рисунок 71 - Схема измерения дифференциальной экспрессии с помощью микроматрицы ДНК
В другом методе используются ДНК-чипы, в которых короткие олигонуклеотиды фотолитографически синтезируются in situ во-время изготовления чипа. Такие батареи ячеек известны как геночипы. Они имеют плотность до 1 000 000 элементов на квадратный сантиметр, причем каждый элемент включает до 109 однонитевых олигонуклеотидов длиной 25 нуклеотидов.
Каждый ген на геночипе представлен двадцатью элементами (двадцатью перекрывающимися олигонуклеотидами); кроме того, для нормализации неспецифической гибридизации в него включены двадцать контрольных несовпадений.
Для скрининга матриц ДНК применяют флуоресцентные РНК-зонды, так как для мечения различных наборов РНК очень удобно использовать разные флуорофоры.
Флуоресцентно меченые РНК-зонды могут быть одновременно гибридизированы на одной матрице, что позволяет проводить непосредственное измерение дифференциальной экспрессии генов.
Гибридизацию геночипов проводят отдельными зондами на двух идентичных чипах, а интенсивности сигналов измеряют и сравнивают с помощью компьютера.
Исходные данные опытов на микроматрице состоят из изображений гибридизированных матриц. Точный характер изображения зависит от подложки матрицы (тип используемой матрицы). Матрицы ДНК могут содержать много тысяч элементов. Поэтому процессы сбора и анализа данных должны быть автоматизированы.
Программное обеспечение для предварительной обработки изображений обычно поставляется вместе со сканером. Оно позволяет определять границы отдельных пятен и измерять полную интенсивность (мощность) сигналов по яркости целых пятен. Интенсивность сигналов необходимо корректировать относительно интенсивности фона, и, кроме того, в матрицу должны быть включены эталоны для измерения неспецифической гибридизации и оценки разброса параметров гибридизации на различных матрицах.
Цель обработки данных состоит в преобразовании сигналов гибридизации в числа, которые могут быть использованы для получения матрицы экспрессии генов. Интерпретация данных гибридизации микроматрицы проводится с помощью их группировки в кластеры согласно подобным профилям экспрессии.
Группировкой называется способ упрощения больших наборов данных за счёт объединения подобных данных в группы (кластеры). Для автоматизации методов анализа данных гибридизации микроматриц были разработаны различные варианты программных приложений, например:
✵ Ресурсы Стэнфордского университета - http://genomics.stanford.edu/
- Stanford Microarray Database (SMD) - http://smd.stanford.edu/
- Microarray Resources: Software and Tools - http://smd.stanford.edu/resources/restech.shtml;
✵ TM4 - http://www.tm4.org/ - Microarray Data Manager (MADAM), TIGR Spotfinder, Microarray Data Analysis System (MIDAS), and Multiexperiment Viewer (MeV), as well as a Minimal Information About a Microarray Experiment (MIAME)-compliant MySQL database;
✵ GeneMaths XT - http://www.applied-maths.com/genemaths-xt;
✵ Eisen Lab - http://rana.lbl.gov/EisenSoftware.htm;
✵ Microarray Image Analysis Software - http://www.statsci.org/micrarra/image.html;
✵ The Gene Ontology - http://www.geneontology.org/GO.tools.microarray.shtml;
✵ BioDiscovery - http://www.biodiscovery.com/ - Nexus Expression - http://www.biodiscovery.com/index/nexus-expression;
✵ BxArrays - http://bioinforx.com/lims/microarray-gene-expression-data-analysis/bxarrays;
✵ Array-Pro Analyzer - http://www.mediacy.com/index.aspx?page=ArrayPro;
✵ Premier Biosoft - http://www.premierbiosoft.com/dnamicroarray/index.html;
✵ J. Craig Venter Institute - http://www.jcvi.org/cms/research/software/;
✵ Bioconductor - http://www.bioconductor.org/.
Микроматрицы ДНК применяют в следующих целях.
1. Исследование состояний клеток и протекающих в них процессов. Анализ дифференциальной экспрессии, зависящей от состояния клетки, помогает в расшифровке механизмов таких процессов, как, например, образование спор или переход от аэробного метаболизма к анаэробному.
2. Диагностика заболеваний. Микроматричный тест на присутствие мутаций может подтвердить диагноз предполагаемого генетического заболевания. Становится возможным обнаружение поздно проявляющихся симптомов, как, например, в случае болезни Хантингтона (см. п. 6.7), и определение потенциально опасных для потомства генов у предполагаемых родителей (рекомендации при планировании семьи).
3. Генетические предупредительные признаки. Некоторые болезни не определяются исключительно генотипом, но вероятность их развития зависит от поведения определённых генов и может быть оценена по картине их экспрессии. Осведомлённый о предрасположении к той или иной болезни, человек в некоторых случаях может предупредить развитие заболевания, изменив свой образ жизни.
4. Подбор лекарственных препаратов. Установление генетических факторов, обусловливающих ответные реакции организма на воздействие медикаментов; у одних пациентов подобные эффекты делают лечение неэффективным, а у других даже вызывают опасные аллергические реакции.
5. Классификация болезней. По разным картинам экспрессии генов могут быть определены, например, различные типы лейкемии. Знание точного типа болезни важно для подбора оптимальных методов лечения.
6. Выбор мишени для разработки лекарства. Белки, показывающие повышенный уровень транскрипции в определённых болезненных состояниях, могли бы быть потенциальными мишенями для фармакологического воздействия (при условии, что по другим данным будет показано, что усиленная транскрипция необходима для поддержания болезненного состояния или способствует ему).
7. Сопротивляемость патогенам. Сравнительный анализ генотипов или картин экспрессии у восприимчивых и стойких к антибиотику бактериальных штаммов позволяет обнаружить белки, вовлечённые в механизм сопротивляемости.
Обнаружение генов. В последнее время значительные денежные средства выделяются на поиск генов, связанных с конкретными видами болезней. Цель этого поиска состоит в развитии новых методов терапии для борьбы с широким спектром распространённых функциональных и структурных расстройств, например, рака, туберкулеза, астмы и т. д.
В настоящее время есть две главные стратегии открытия белков, которые могут представлять собой молекулярные мишени, подходящие как для получения молекулярных препаратов, так и для развития гено-терапии.
Одним из подходов к обнаружению связанных с болезнью генов является метод позиционного клонирования. Согласно этому методу изучают популяцию людей, в которой наблюдаются случаи рассматриваемого заболевания, и находят хромосому, связанную с развитием этой болезни.
Затем устанавливают связь болезни с некоторой хромосомной областью, после чего секвенируют большой отрезок хромосомы вблизи этой области (локуса) и получают последовательность ДНК длиной несколько сот тысяч пар нуклеотидов. В принципе такой локус может содержать множество генов, хотя, скорее всего, только один из них действительно вовлечён (прямо или косвенно) в болезнетворный процесс.
Для повышения эффективности распознавания генов в локусе могут быть использованы различные методы поиска последовательностей и предсказания генов, но так или иначе должны быть экспрессированы несколько генов, и для установления того, какой именно ген действительно вовлечён в болезнь, потребуется дальнейший анализ (или испытания).
Хотя гены, обнаруженные этим способом, могут быть вполне удовлетворительными с академической точки зрения, они вовсе не обязательно будут хорошими мишенями для лекарственных препаратов (или точками терапевтического воздействия).
Другой подход к открытию генов, требующий намного меньших затрат на секвенирование и больше полагающийся на мощные поисковые возможности современных вычислительных систем, основан на отыскании генов, которые фактически экспрессируются в здоровых и больных тканях. Он позволяет проводить сравнение уровней экспрессии в двух состояниях и на основании такого анализа наиболее эффективно выбирать потенциальную мишень. Этот процесс анализирует те молекулы мРНК, которые используются для синтеза этих белков.
Как правило, в обнаружении генов участвуют следующие элементы:
✵ участки сращения;
✵ старт- и стоп-кодоны;
✵ точки разветвления;
✵ промоторы и терминаторы транскрипции;
✵ участки полиаденилирования;
✵ сайты связывания рибосом;
✵ сайты связывания с топоизомеразой II;
✵ сайты расщепления топоизомеразой I;
✵ сайты связывания с различными факторами транскрипции.
Такие локальные участки называют "сигналами" и обнаруживают с помощью "датчиков сигналов". Напротив, удлинённые последовательности и последовательности переменной длины (например, экзонов и интронов) называют "содержанием" и обнаруживают посредством "датчиков содержания".
Наиболее сложные из применяемых датчиков сигналов - нейросети.
Типичный датчик содержания - тот, который предсказывает кодирующие области.
Для определения полной структуры гена было создано несколько систем, комбинирующих датчики сигналов и содержания. Такие системы способны распознавать более сложные взаимозависимости между свойствами генов. Одной из первых комплексных программ поиска генов, разработанных на сегодняшнее время, является программа Genelang (http://arete.ibb.waw.pl/PL/html/gene_lang.html). Построенная на алгоритмах динамического программирования, эта программа комбинирует отобранные экзоны и другие области или участки с назначаемым счётом и предсказывает целый ген с максимальным полным счётом.
При геномных исследованиях активно используются скрытые марковские модели (Hidden Markov model, НММ) (см. п. 9.3). Марковские модели, реализованы, например, в программах:
✵ Genemark - http://exon.biology.gatech.edu/ и http://opal.biology.gatech.edu/GeneMark/gmhmm2_prok.cgi;
✵ GlimmerM - http://www.cbcb.umd.edu/software/glimmerm/;
✵ Critica - http://www.ttaxus.com/soflware.html;
✵ AMIGene - http://www.genoscope.cns.fr/agc/tools/amigene/Form/form.php;
✵ EasyGene - http://www.cbs.dtu.dk/services/EasyGene/.
У прокариотов локус гена всё ещё принято определять путём тривиального поиска открытой рамки считывания. Такой способ, конечно, не пригоден для высших эукариотов.
Для разделения кодирующих и некодирующих областей у высших эукариотов применяют датчики содержания экзонов, построенные на статистических моделях частот использования нуклеотидов и проводящие статистическую оценку некоторых зависимостей, наблюдаемых в структуре кодона.
Профили экспрессии генов. Геном человека невероятно сложен и состоит приблизительно из трёх миллиардов пар нуклеотидов ДНК. При этом лишь только 3% ДНК является кодирующей последовательностью (то есть той частью генома, которая транскрибируется в РНК и затем транслируется в белок).
Остальная часть генома состоит из областей, необходимых для компактного хранения хромосом, их репликации во время деления клетки, управления транскрипцией и т. д.
Основная часть работы по анализу последовательности генома приходится на исследование продуктов клеточных механизмов транскрипции и трансляции, то есть на анализ белковых последовательностей и структур.
В последнее время значительные усилия направлены на автоматизацию процессов исследования мРНК; частично это связано с тем, что смысловая машинная трансляция мРНК в последовательность белка может быть легко реализована алгоритмически, но главная причина состоит в том, что молекулы мРНК представляют ту часть генома, которая экспрессируется в клетках определенного типа на определенном этапе их развития.
Таким образом, можно выделить три уровня геномной информации:
1) геном хромосом (собственно геном) - генетическая информация, общая для всех клеток организма;
2) экспрессируемый геном (транскриптом) — та часть генома, которая экспрессируется в клетке на определённой стадии её развития;
3) протеом - совокупность белковых молекул, взаимодействие которых придаёт клетке её индивидуальные качества.
Каждый уровень требует различных аналитических методов и объяснительных алгоритмов. На разных стадиях развития и уровнях биологической активности клетки экспрессируют различный набор генов.
Такой характеристический набор экспрессируемых генов называют профилем экспрессии этой клетки.
Зарегистрировав профили экспрессии некоторой клетки, можно воссоздать картину уровней экспрессии генов в нормальном или ненормальном состоянии клетки, а также картину относительных уровней экспрессии всех генов, транскрибируемых в этой клетке. Кроме того, анализ зарегистрированных профилей экспрессии позволяет обнаруживать новые гены, тем самым дополняя другие методы, используемые в глобальных проектах секвенирования генома.