Молекулярная биология: Структура и функции белков - Степанов В.М. 2005
Первичная структура белка
Методы определения первичной структуры
4.4.1. Подготовка белка к анализу
Подготовка белка к анализу первичной структуры призвана свести к минимуму влияние других, более высоких уровней его организации. Иными словами, объектом анализа должна быть неупорядоченная полипептидная цепь без каких-либо поперечных ковалентных связей (например, дисульфидных мостиков) так, чтобы все ее звенья, все пептидные связи были в равной мере доступны действию как химических реагентов, так и ферментов. К сожалению, в полной мере исключить помехи со стороны свойственных белку нековалентных взаимодействий не удается, однако разработаны приемы, позволяющие приблизиться к этой цели.
Итак, белок должен быть прежде всего подвергнут глубокой денатурации и утратить четвертичную, третичную и по возможности вторичную структуру. Если в нем имеются дисулъфидные связи, прибегают к их расщеплению, используя, как правило, восстановление большим избытком меркаптосоединения, например дитиоэритрита или меркаптоэтанола:
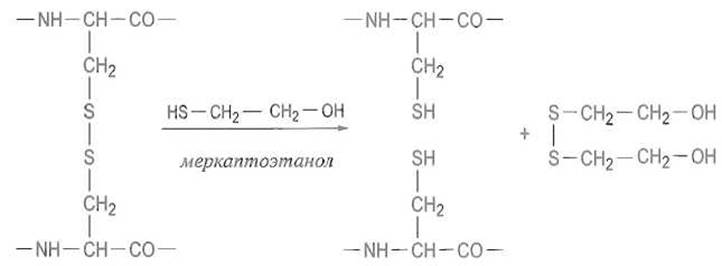
Эта реакция, которую обычно проводят в условиях, обеспечивающих денатурацию белка (в 8 М растворе мочевины или в 6 М гидрохлориде гуанидина при pH 8-9), имеет своим результатом превращение остатка цистина в два остатка цистеина со свободны ми сульфгидрильными группами. Сохранять такое производное и тем более использовать его для последующих превращений нельзя, так как после удаления избытка восстановителя, что обычно достигается гель-фильтрацией смеси, вновь произойдет замыкание дисульфидных связей за счет окисления свободных SH-групп кислородом воздуха. Во избежание этого сульфгидрильные группы блокируют, обрабатывая восстановленный и денатурированный белок избытком иодуксусной кислоты, которая переводит остатки цистеина в устойчивый S-карбоксиметилцистеин:

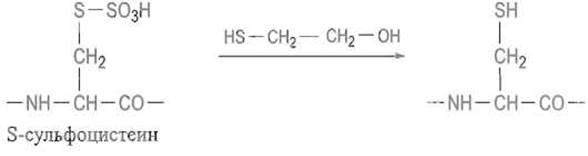
Такое превращение необратимо. Если желательна последующая регенерация остатков цистеина, прибегают к получению S-сульфопроизводных, обрабатывая белок сульфитом натрия в присутствии ионов меди. Получающийся при этом S-сульфоцистеин при действии избытка меркаптосоединений количественно переходит в цистеин:
Несмотря на совершенствование автоматических методов определения: первичной структуры, даже в благоприятных случаях редко удается в одной цепи за один прием определить последовательность более чем 50 аминокислотных остатков. Этого достаточно для исследования небольших белков, в остальных же случаях необходимо прибегать к фрагментации полипептидной цепи несколькими методами, с тем чтобы получить систему перекрывающихся пептидов. Определение их строения и сопоставление полученных данных приводят к установлению последовательности аминокислот всей полипептидной цепи белка.
Известны ферментативные и химические методы специфического расщепления полипептидной цепи.
4.4.2. Ферментативные методы фрагментации полипептидной цепи
При выборе протеолитических ферментов, подходящих для избирательного гидролиза белка, особенно важна их специфичность. Как правило, пользуются так называемой первичной специфичностью протеиназ, т.е. их способностью преимущественно атаковать пептидные связи, образованные тем или иным аминокислотным остатком. Аминокислотные остатки субстрата принято обозначать латинской буквой Р с индексами, соответствующими номеру остатка, считая от расщепляемой связи, к аминному концу («влево»). Остатки, расположенные в сторону карбоксильного конца («вправо» от гидролизуемой связи), обозначают Р' с соответствующим индексом:
...Р4 -Р3 -Р2 -P1 -СО-NH-P'1 -P'2 -Р'3.
Если использовать эти обозначения, то первичной специфичностью следует считать способность той или иной протеиназы избирательно расщеплять пептидные связи, образованные остатками Р1 и Р'1. Все протеиназы, однако, имеют более или менее выраженную вторичную специфичность: скорость расщепления ими пептидной связи зависит не только от строения той аминокислоты, которая прямо участвует в ее построении своей карбоксильной или аминной группой (т.е. остатка P1 и P'1), но и от соседних аминокислотных остатков Р2,Р2,Р'2Р'3 и т.д. Обычно в образовании комплекса с ферментом участвует не менее 5-6 аминокислотных остатков (см. гл. 10).
Влияние вторичной специфичности уменьшают, проводя гидролиз достаточно долго, так, чтобы расщепились и те пептидные связи, которые находятся в невыгодном для данного фермента окружении. Специфичность протеиназ редко бывает совершенно строгой: наблюдается и расщепление нехарактерных для данного фермента связей, нарастающее со временем. Все это требует оптимизации условий ферментативного гидролиза полипептидных цепей.
В настоящее время для специфического гидролиза белков при определении первичной структуры наиболее часто применяют следующие протеиназы.
Трипсин. Это фермент поджелудочной железы животных, который принадлежит классу так называемых сериновых протеиназ и специфически гидролизует пептидные связи, образованные карбоксильными группами остатков аргинина или лизина (P1 = Arg, Lys). Боковые цепи этих аминокислот несут на определенном расстоянии от а-углеродного атома положительно заряженную катионную группу. Показано, что в зоне связывания субстрата последняя входит во впадину, где расположена отрицательно заряженная ß-карбоксильная группа одного из остатков аспарагиновой кислоты трипсина:
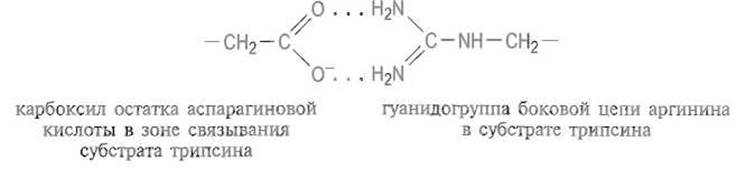
Электростатическое взаимодействие между катионной и анионной группами играет определяющую роль в специфичности трипсина. Это подтверждается тем, что замена аспарагиновой кислоты в зоне связывания субстрата другим остатком приводит к утрате способности гидролизовать пептидные связи аргинина или лизина.
Блокирование е-аминогрупп лизина в атакуемом трипсином полипептиде, например ацилированием цитраконовым ангидридом (см. гд 9), приводит к замене катионной аминогруппы на анионную (карбоксилатную), поэтому в соответствующем производном трипсин расщепляет только пептидные связи аргинина. Это позволяет получить более крупные фрагменты белка, которые после их выделения из гидролизата выдерживают в слабокислом растворе, что вызывает отщепление остатка цитраконовой кислоты и высвобождение е-аминогрупп лизина. Затем такие пептиды с регенерированными остатками лизина вновь расщепляют трипсином.
Аналогично можно регулировать доступность пептидных связей аргинина гидролизу трипсином. Для этого блокируют гуанидо группу аргинина, обрабатывая белок в щелочной среде дикетоном, например диацетилом, закрепляя образующийся при этом диол образованием комплекса с борной кислотой (см. гл. 9).
При гидролизе белка трипсином обычно используют pH, близкий 8, температуру 37°С и весовое соотношение фермент — субстрат 1:100, регулируя полноту гидролиза временем реакции. Нередко при гидролизе трипсином расщепляются отдельные пептидные связи, соответствующие специфичности родственного фермента — химотрипсина. Это может объясняться присутствием в препарате трипсина примеси химотрипсина или продуктов ограниченного гидролиза трипсина, однако и вполне чистый трипсин может сам по себе обнаруживать нехарактерную для него специфичность.
Химотрипсин. Сериновая протеиназа поджелудочной железы, структурно родственная (гомологичная) трипсину. Химотрипсин специфически гидролизует (оптимум pH вблизи 8) пептидные связи, образованные карбоксильными группами ароматических аминокислот — фенилаланина, тирозина, триптофана и нередко лейцина, сравнимого с этими остатками по гидрофобности (Р1 = Phe, Tyr, Trp, Leu).
Термолизин. Металлопротеиназа, секретируемая клетками термофильной бактерии Bacillus amyloliquefaciens. Этот фермент, имеющий ион цинка в активном центре, оптимально активен при pH 6-8 и избирательно гидролизует пептидные связи, образованные с участием аминогрупп гидрофобных аминокислот — лейцина, фенилаланина, тирозина, валина, изолейцина (P'1 = Leu, Phe, Tyr, Val, Ile). Фермент может применяться даже при 60-80°С.
Glu, Asp-специфичные протеиназы (эндопротеиназы Glu—С). Сериновые протеиназы Staphylococcus aureus, а также некоторых стрептомицетов и термоактиномицетов. Ферменты гидролизуют пептидные связи, образованные а-карбоксильными группами остатков глутаминовой кислоты, значительно реже аспарагиновой кислоты (P1 = Glu, Asp). Имеют в зависимости от субстрата и состава буферного раствора два оптимума pH: при 4 и 8. Характерная для этих протеиназ и весьма строго выдерживаемая специфичность хорошо дополняет специфичность других протеолитических ферментов, поэтому они все шире применяются в анализе структуры.
Эндопротеиназа Lys-C. Протеолитический фермент из Lysobacter enzymogenes, специфически гидролизующий пептидные связи, образованные карбоксильной группой лизина, но не аргинина (P1= Lys).
Эвдопротеиназа Arg-C. Протеиназа из подчелюстной железы мыши, избирательно расщепляющая пептидные связи, образованные с участием карбоксильной группы аргинина (P1 = Arg).
Можно предполагать, что будут найдены и войдут в практику фрагментации белков и другие протеолитические ферменты с четко определенной специфичностью.
4.4.3. Химические методы специфического расщепления пептидных связей
Из множества предлагавшихся в различное время методов избирательного химического расщепления пептидных связей наибольшее распространение в практике анализа первичной структуры получило расщепление бромцианом, проводимое обычно в муравьиной или трифторуксусной кислоте. Эта реакция ведет к расщеплению пептидных связей, образованных с участием карбоксильной группы остатков метионина. Бромциан реагирует с серой тиоэфирной группы метионина, образуя сульфониевую соль. В этом промежуточном продукте положительный заряд на атоме серы индуцирует возникновение частичного положительного заряда (δ+) на соседнем у-атоме углерода боковой цепи метионина. Возникший таким образом на у-углероде электрофильный центр подвергается атаке кислородным атомом карбонильной группы, на котором постоянно существует частичный отрицательный заряд, усиливающийся во время реакции, что обусловлено поляризацией связи С=0.
В результате смещение электронной плотности в этой группе и во всей пептидной связи завершается образованием ковалентной связи между у-углеродным атомом и атомом кислорода, двойная связь С=0 переходит в простую, а связь С—N пептидной группы, обычно частично двойная, становится полностью двойной с переносом положительного заряда на ее атом азота:

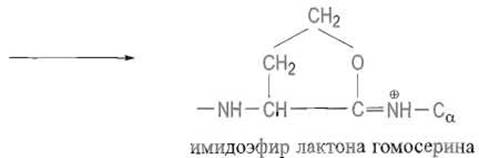
Одновременный разрыв связи S—СН2 освобождает летучий метилроданид. Получившееся в результате внутримолекулярной реакции производное имидоэфира лаптопа гомосерина легко гидролизуется с разрывом связи С—N, что и завершает специфическое расщепление пептидной связи остатка метионина. На С-конце вновь образовавшегося пептидного фрагмента оказывается остаток гомосерина или его лактон, легко превращающиеся друг в друга:

Метод расщепления бромцианом достаточно избирателен, затруднения встречаются лишь в тех случаях, когда за остатком метионина следуют серин или треонин. Характерно, что к расщеплению пептидных связей бромцианом прибегают при «вырезании» из гибридных генноинженерных конструкций пептидов или небольших белков, встроенных в структуру белка-хозяина для лучшей экспрессии в бактериальных клетках.
Среди других методов химического расщепления пептидной связи следует упомянуть более или менее избирательное выщепление остатков аспарагиновой кислоты при кратком нагревании белка или пептида в слабокислом растворе. Видимо, внутримолекулярный катализ с участием ß-карбоксильной группы аспарагиновой кислоты, которая может быть сближена с обеими пептидными связями, образованными этим остатком, ответствен за преимущественный гидролиз именно этих связей. В еще более мягких условиях при выдерживании в кислых растворах избирательно расщепляются пептидные связи Asp—Pro.
Описаны также методы специфического расщепления пептидных связей триптофана, цистеина и пролина, однако они нашли лишь весьма ограниченное применение.
4.4.4. Разделение пептидов, полученных фрагментацией полипептидной цепи
Разделение смесей пептидов, получаемых в результате ферментативного или химического расщепления полипептидной цепи белка, составляет одну из наиболее сложных, нестандартных, стадий анализа первичной структуры. Традиционная схема разделения состояла из большого числа стадий и, следовательно, была сопряжена с очень большими потерями пептидов, причем ведущую роль играла ионообменная хроматография пептидов чаще всего на сульфополистирольных катионитах с элюцией в градиенте pH. Метод дополняли гель-фильтрацией, хроматографией и электрофорезом на бумаге или тонкослойной хроматографией.
В последние годы доминирует фракционирование пептидов высокоэффективной жидкостной хроматографией (ВЭЖХ) на сорбентах с «обращенной фазой». Последние представляют собой мелкие частицы кремнезема с большой поверхностью, к которой ковалентно присоединены углеводородные цепи различной длины. Разделение на таких сорбентах определяется гидрофобными взаимодействиями между этими цепями, содержащими 4, 8, иногда 18 углеродных атомов (C4, C8, C18), и гидрофобными элементами в структуре пептидов. Определенную роль играют и ионообменные эффекты. Пептиды элюируют при высоких давлениях — порядка 100 атм, используя градиент концентрации органических растворителей с различной десорбирующей способностью (метанол, ацетонитрил, изопропанол, диоксан), к которым для регулирования pH прибавляют трифторуксусную кислоту или буферные смеси.
Хорошие результаты дает также ионообменная хроматография на мелкозернистых ионитах типа Моно-Q и Moho-S (см. гл. 3) с использованием давления порядка 20 атм. Эти методы, обладающие высокой разрешающей способностью, позволили резко сократить число стадий выделения индивидуальных пептидов, снизить их потери и перейти к анализу первичной структуры очень малых (порядка сотен наномоль) количеств белка.