Молекулярная биология: Структура и функции белков - Степанов В.М. 2005
Пептиды
Химический синтез пептидов
Синтез пептидов, основы которого были заложены Э. Фишером в начале XX в., развился в обширную область синтетической органической химии. Описаны синтезы весьма протяженных пептидов и нескольких, пока небольших, белков. Не умаляя значения последних работ, укажем, что для практического получения значительных количеств белка предпочтителен микробиологический синтез в рекомбинантных клетках; начал развиваться и биосинтез пептидов в бесклеточных системах.
Синтез пептидов определенной структуры, даже наиболее простых, требует предварительного временного блокирования (защиты) функциональных групп, которые не должны участвовать в реакции. Если этого не предусмотреть, будут получаться весьма сложные смеси продуктов.
2.2.1. Защита аминогруппы
а-Аминогруппу ацилирующей аминокислоты или пептида защищают так, чтобы блокирующую группировку после образования пептидной связи можно было отщепить, снять в достаточно мягких условиях, которые гарантировали бы целостность пептида. Одной из первых была предложена (сохранившая значение до сих пор) бензилоксикарбонилъная группа (сокращенное обозначение Z, устаревшее название карбобензоксигруппа), которую вводят, ацилируя аминокислоту или пептид бензилоксикарбонилхлоридом:
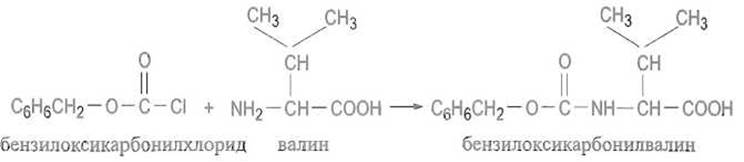
Если надобность в защите миновала, бензилоксикарбонильную группу отщепляют гидрированием в присутствии палладиевого катализатора или действием бромистого водорода в ледяной уксус ной кислоте:
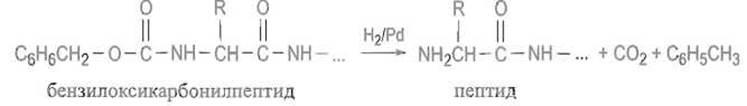
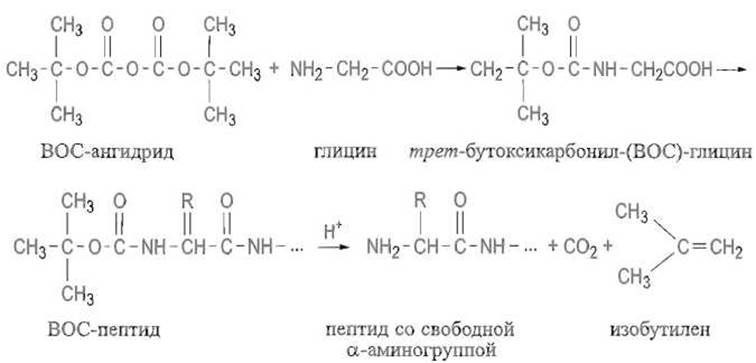
Широко применяют трет - бутоксикаобонильную (ВОС) группу. Ее вводят, ацилируя аминогруппу ангидридом моно-трет-бутилового эфира угольной кислоты (ВОС-ангидридом), отщепление же достигается мягким действием кислотных агентов — трифторуксусной кислоты или хлористого водорода в органических растворителях:
В последнее время, особенно в автоматическом синтезе пептидов, все чаще используют 9-флуоренилметилоксикарбонильную группу (Fmoc-группу). Ее вводят, действуя на аминокислоту или пептид соответствующим хлорангидридом:

* Вызывает элиминирование водорода, указанного стрелкой.

Известно множество других защитных групп, обладающих теми или иными преимуществами и недостатками и применяемых в различных схемах синтеза. Необходимость использования большого их набора обусловлена тем, что специфические группы требуются для защиты функциональных групп боковых цепей, причем деблокирование а-аминогрупп необходимо проводить каждый раз перед новой стадией синтеза, тогда как защита боковых групп не должна затрагиваться в этих условиях. Их отщепляют в самом конце синтеза пептида.
2.2.2. Защита а-карбоксильной группы
Для этой цели обычно используют превращение карбоксильной группы в метиловый или трет-бутиловый эфир по реакциям, описанным в гл. 1. Для отщепления метилового эфира прибегают к его омылению щелочью, которое не всегда протекает гладко и создает опасность рацемизации С-концевого остатка. Трет-бутловые эфиры удобно расщеплять мягкой обработкой кислотными агентами, например трифторуксусной кислотой:

2.2.3. Образование пептидной связи
Для образования пептидной связи, которое при химическом синтезе, как правило, проводят в отсутствие воды, необходима активация карбоксильной группы. Возможна, впрочем, и активация а-аминогруппы, однако ее практически не применяют. Активированное производное защищенной по аминогруппе аминокислоты или пептида может быть получено в отдельной реакции, а иногда и выделено или образовано непосредственно перед конденсацией с аминокомпонентом (аминокислотой или пептидом с защищенной карбоксильной группой).
Реакция в присутствии карбодиимидов. Ацилированная аминокислота реагирует с —N =С =N— (карбодиимидной) группировкой, образуя соответствующую о-ацилмочевину. Это активированное производное вступает в реакцию с аминокомпонентом, причем образуются пептид и двузамещенная мочевина:
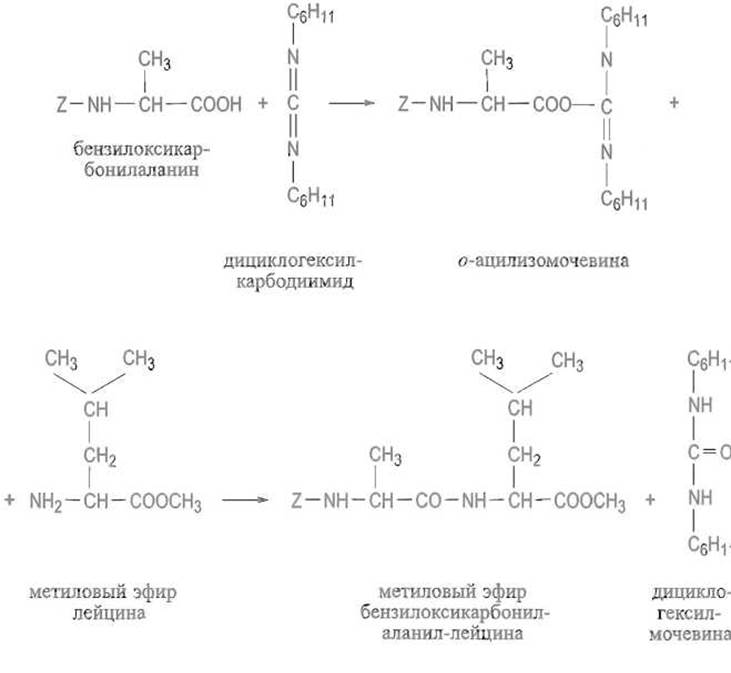
Активированные эфиры. Иногда удобнее превратить о-ацилмочевину в активированный эфир (с некоторым допущением и само это соединение можно рассматривать как активированный эфир; однако оно нестабильно, что может привести к нежелательным побочным реакциям). Для этого к реакционной смеси, содержа щей o-ацилмочевину, прибавляют пентафторфенол, n-нитрофенол, N-оксисукцинимид и т.п.:
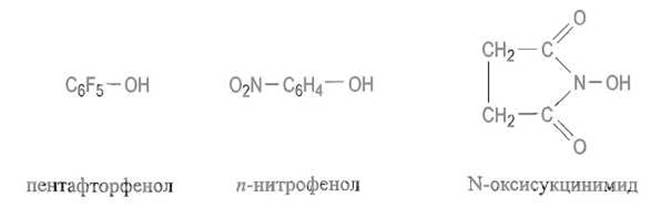
Продукты переноса аминоацильного или пептидного остатка на оксигруппу этих соединений относительно устойчивы и могут быть выделены. Их взаимодействие с аминокомпонентом дает требуемый пептид:

Симметричные ангидриды защищенных аминокислот. o-Ацил-мочевина способна реагировать с карбоксильной группой второй молекулы защищенной аминокислоты, образуя ее симметричный ангидрид. Последний является хорошим ацилирующим агентом и реагирует с аминокомпонентом, в результате чего получается пептид и отщепляется защищенная аминокислота:
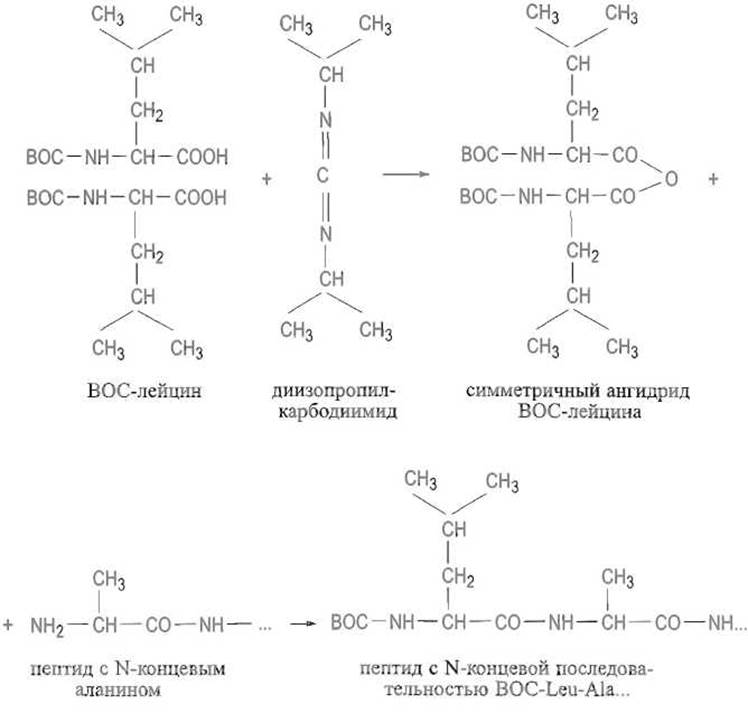
Известно и применяется гораздо большее число способов активации карбоксильных групп; их описание можно найти в специальных руководствах.
2.2.4. Тактика пептидного синтеза
Изложенные подходы позволяют проводить направленный синтез весьма сложных пептидов. Оптимальный путь синтеза выбирают исходя из ряда соображений. Особое внимание уделяют предотвращению рацемизации, наиболее вероятной у Са-атома того остатка, карбоксильная группа которого активирована. Это обусловлено тем, что активирующие группы вызывают смещение электронной плотности не только от углерода карбонильной группы (что необходимо для облегчения атаки этого атома нуклеофильной аминогруппой аминокомпонента), но и от С а-атома. Последнее благоприятствует отщеплению атома водорода, находящегося при этом углероде, в виде протона. Такое отщепление переводит остающиеся три заместителя у а-атома углерода в плоскую тригональную конфигурацию. При возврате протона, который может подойти к Са-атому с обеих сторон плоскости, происходит образование равной смеси L- и D-изомеров, т е. рацемизация. Таким образом, активация карбоксильной группы несет в себе потенциальную опасность рацемизации С-концевых остатков аминокислот в пептидных фрагментах:
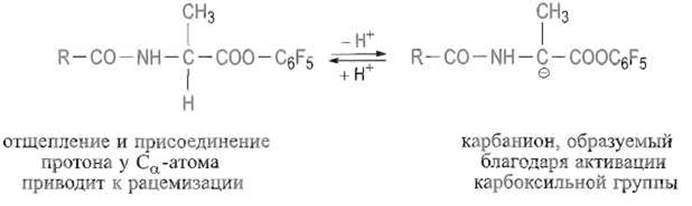
Амидные связи уретанового типа в бензилоксикарбонил- и ВОС-аминокислотах менее благоприятствуют рацемизации по сравнению с обычными пептидными связями. Следовательно, планируя синтез пептида, выгодно не конденсировать его фрагменты, поскольку для этого пришлось бы активировать карбоксильные группы пептидов, а присоединять к растущему пептиду защищенные активированные аминокислоты одну за другой начиная с С-концевого аминокислотного остатка. Синтез длинных пептидов иногда приходится все же проводить из фрагментов, которые затем соединяют друг с другом. В этом случае стараются выбирать фрагменты так, чтобы в С- концевом положении каждого из них оказывался остаток глицина, который не содержит асимметрического атома и, значит, не может рацемизоваться, или остаток пролина, в котором нет атома водорода при а-утлеродном атоме и не может происходить рацемизация по описанному выше механизму. Если такой прием почему-либо невозможен, прибегают к конденсации фрагментов, используя азиды пептидов, — метод, при котором опасность рацемизации минимальна, хотя и не исключена вовсе.
Исключительно ценным развитием ступенчатого синтеза пептидов явился твердофазный метод синтеза пептидов, предложенный Р. Меррифилдом. С-концевую аминокислоту присоединяют к полимерной матрице сложноэфирной связью, используя, например, реакцию соли аминокислоты с активными бромметильными группами на поверхности поперечно сшитого полистирола:

После образования сложноэфирной связи между С-концевым остатком и матрицей отщепляют защитную группу (обычно ВОС или Fmoc), обрабатывая полимер соответственно кислотой или амином:
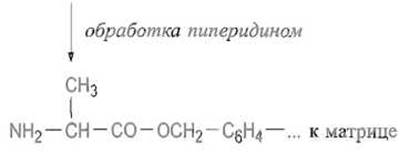
К освободившейся а-аминогруппе присоединяют аминокислоту, предшествующую С-концевой, используя для этого ангидрид защищенной аминокислоты или иной способ активации — реакцию в присутствии карбодиимида, пентафторфенилового эфира защищенной аминокислоты и т п:

Затем снова отщепляют защитную группу и повторяют цикл, присоединяя следующий аминокислотный остаток, и т.д.
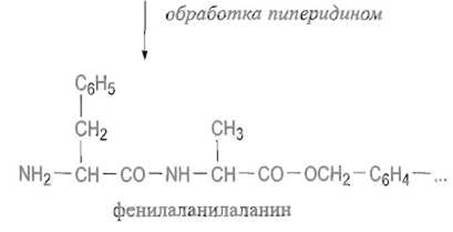
Такие циклы можно повторять много раз, получая весьма длинные пептидные цепи, содержащие до 100 остатков и более. Многостадийный синтез на твердофазном носителе выгоден прежде всего тем, что он позволяет избежать потерь синтезируемого пептида, неминуемых при выделении продукта синтеза в обычных: условиях. Выход на стадии присоединения аминокислоты и отщепления защитной группы достигает, как правило, 99-99,5%, в частности за счет применения большого избытка ацилирующего компонента. Важно и то, что растущие пептидные цепи, будучи связаны с носителем, защищены от агрегации — явления, резко ограничивающего возможности синтеза длинных пептидов в растворе.
Твердофазный синтез пептидов, который состоит из строго одинаковых повторяющихся стадий, удалось автоматизировать — были созданы автоматические синтезаторы пептидов.
На практике метод все же ограничен неполнотой присоединения ациламинокислоты в отдельных циклах. Понятно, что «недостроенный» в том или ином цикле пептид будет ацилирован в следующем, что приведет к пропуску аминокислотного остатка. Образование таких пептидов с ошибочными последовательностями даст при синтезе длинного пептида весьма сложную смесь продуктов. Метод оказался особенно продуктивным в сочетании с высокоэффективной жидкостной хроматографией, которая позволяет выделить главный компонент среди множества продуктов твердофазного синтеза.
Сочетание твердофазного синтеза и высокоэффективной жидкостной хроматографии в настоящее время — главный способ получения небольших количеств 15-20-членных пептидов, в частности пептидных детерминант, используемых для получения специфических антител к тому или иному белку.
Применение различных приемов пептидного синтеза, по преимуществу твердофазного, позволило получить некоторые, как правило, не очень большие белки. Первоначально эти работы имели только принципиальный характер, демонстрируя возможность чисто химического синтеза белков. В последнее время, однако, проведены синтезы белков, имеющие практическое значение. В частности, в значительных количествах удается получать инсулин, ряд других пептидных гормонов, например кальцитонин рыб (32 аминокислотных остатка), синтезируемый партиями по 50—100 г, фактор роста эпителия (53 остатка), росттрансформирующий фактор а (50 остатков), интерлейкин-3 (140 остатков), a1- и а2-интерфероны лейкоцитов человека и их аналоги, панкреатический ингибитор трипсина. Синтезирована и использована для определения пространственной структуры протеиназа вируса иммунодефицита человека, пептидная цепь которой содержит 99 аминокислот. Характерно, что свертывание полученных синтетически пептидных цепей в нативную структуру происходит самопроизвольно и не вызывает существенных затруднений.
Несмотря на то что получение белков в значительных количествах в ближайшем будущем будет, по-видимому, основано на микробиологическом синтезе или синтезе в бесклеточной системе, возможны и дальнейшие успехи в развитии химических способов получения крупных пептидов и белков. Определенные перспективы имеет и так называемый полусинтез белков, при котором полипептидную цепь белка собирают из пептидных фрагментов, полученных ограниченным протеолизом и чисто химическим путем с использованием химических, а иногда энзиматических методов их конденсации. Такой подход был применен, в частности, для получения проинсулина человека присоединением ряда синтетических пептидных звеньев к выделенным из природного инсулина А- и В-цепям.